
CASIRIVIMAB et IMDEVIMAB 120 mg-mL, solution à diluer pour perfusion intraveineuse ou solution pour injection sous-cutanée, boîte de 2 flacons unidoses de 2,50 ml
Retiré du marché le : 03/07/2023
Dernière révision : 30/11/2021
Taux de TVA : 0%
Laboratoire exploitant : ROCHE
Source :

Traitement de la COVID-19 :
L'association casirivimab et imdevimab est indiquée dans le traitement de la COVID-19 confirmée par un test virologique de détection du SARS-CoV-2 positif, chez les patients âgés de 12 ans et plus :
- ne nécessitant pas d'oxygénothérapie du fait de la COVID-19: le traitement doit être instauré dès que possible après l'obtention du test RT-PCR au SARS-CoV-2 positif et dans un délai maximum de 5 jours après le début des symptômes ;
- hospitalisés du fait de la COVID-19 et séronégatifs (IgG anti-Spike) nécessitant une oxygénothérapie non invasive ;
ET étant à risque élevé d'évolution vers une forme grave de la maladie à savoir les populations suivantes telles que définies par l'ANRS-Maladies Infectieuses Emergentes:
· Les patients ayant un déficit de l'immunité lié à une pathologie ou à des traitements :
- Chimiothérapie en cours
- Transplantation d'organe solide
- Allogreffe de cellules souches hématopoïétiques
- Maladie rénale avec DFG <30 mL/min ou dialyse
- Lupus systémique ou vascularite avec traitement immunosuppresseur
- Traitement par corticoïde >10 mg/jour d'équivalent prednisone pendant plus de 2 semaines
- Traitement immunosuppresseur incluant rituximab
- Infection par le VIH non contrôlée ou stade SIDA
· Les patients à risque de complications:
- Obésité (IMC>30),
- BPCO et insuffisance respiratoire chronique,
- Hypertension artérielle compliquée,
- Insuffisance cardiaque,
- Diabète (de type 1 et de type 2),
- Insuffisance rénale chronique,
- Fibrose pulmonaire idiopathique
- Sclérose latérale amyotrophique
- Pathologies rares du foie y compris hépatites auto-immunes
- Myopathies avec capacité vitale forcée <70%
- Autres pathologies rares définies par les filières de santé maladies rares (FSMR)
- Trisomie 21
· Les patients de 80 ans et plus
Prévention de la COVID-19 :
L'association casirivimab et imdevimab est indiquée :
· en prophylaxie pré-exposition de l'infection à SARS-CoV-2 chez les patients adultes et les enfants âgés de 12 ans et plus, n'ayant pas développé du fait de leur immunodépression une réponse vaccinale après un schéma vaccinal complet conformément aux recommandations vaccinales en vigueur1 (patients non répondeurs2) ;
· en prophylaxie post-exposition de l'infection à SARS-CoV-2 chez les patients adultes et les enfants âgés de 12 ans et plus, n'ayant pas développé du fait de leur immunodépression une réponse vaccinale satisfaisante après un schéma vaccinal complet conformément aux recommandations vaccinales en vigueur (patients non répondeurs ou faiblement répondeurs3)
ET appartenant à l'un des sous-groupes à très haut risque de forme sévère de COVID-19 tels que définis par l'ANRS-Maladies Infectieuses Emergentes :
- Receveurs de greffes d'organes solides,
- Receveurs d'une greffe allogénique de cellules souches hématopoïétiques,
- Hémopathies lymphoïdes : leucémies lymphoïdes chroniques traitées ou non, lymphomes non hodgkiniens et myélomes sous traitement, y compris les patients receveurs de thérapie cellulaire génique de type CAR-T cell ou d'anticorps thérapeutiques bi-phénotypiques,
1 Ministère des Solidarités et de la Santé. DGS. 2021-DGS-Urgent 61 : Evolutions diverses de la campagne vaccinale. Disponible sur : https://solidarites-sante.gouv.fr/professionnels/article/dgs-urgent [Consulté le 02/08/2021]
2 Sont considérés comme non répondeurs les patients dont la concentration ou le titre en anticorps anti-S est inférieur au seuil
de positivité défini par le fabricant. Si le test sérologique présente une zone grise définie par le fabricant, les patients présentant une concentration ou un titre d'anticorps anti-S compris dans cette zone sont également considérés comme non répondeurs
3 Patient faiblement répondeur défini par un titre d'anticorps anti-S compris entre la zone grise et 260 BAU/mL et après un schéma vaccinal complet et conformément aux recommandations (comprenant au moins 3 doses de vaccin anti-SARS-CoV-2)
- Patients recevant un traitement par anticorps anti-CD20 ou inhibiteurs de BTK ou azathioprine, cyclophosphamide et mycophenolate mofetil,
- Sujets porteurs d'un déficit immunitaire primitif.
Oulespatients 4 séronégatifs après un schéma vaccinal complet ou non éligibles à la vaccination etqui présentent une immunodépression sévère etqui sont à haut risque de forme sévère de la COVID-19.
Dans le cadre d'une administration en prophylaxie pré-exposition, le casirivimab et l'imdevimab doivent être administrés simultanément et de façon répétée toutes les 4 semaines dès lors qu'il existe un risque d'être exposé au SARS-CoV-2.
· Les patients doivent avoir la confirmation d'un test RT-PCR négatif avant chaque administration.
Dans le cadre d'une administration en prophylaxie post-exposition, le casirivimab et l'imdevimab doivent être administrés simultanément dès que possible après l'exposition confirmée au SARS-CoV-2.
· Les sujets cas-contact doivent avoir la confirmation d'un test RT-PCR négatif avant l'administration ;
· En contexte d'urgence, les patients tels que définis ci-dessus n'ayant pas reçu un schéma vaccinal complet ou avec une exposition à un patient COVID-19 dans les 7 jours après la dernière dose, peuvent également bénéficier de la prophylaxie post-exposition sans attendre le résultat de la sérologie.
L'association casirivimab et imdevimab n'est pas destinée à être utilisée comme substitut de la vaccination contre le SARS-CoV-2.
Cette indication est susceptible d'évoluer en fonction de l'état des connaissances scientifiques et du contexte épidémiologique.
Hypersensibilité au casirivimab ou à l'imdevimab ou à l'un des excipients mentionnés dans la rubrique Liste des excipients.
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom de spécialité et le numéro de lot du produit administré doivent être clairement enregistrés.
Réactions d'hypersensibilité notamment réactions liées à la perfusion
Des réactions graves d'hypersensibilité incluant des réactions anaphylactiques pouvant mettre en jeu le pronostic vital ont été observées avec les anticorps monoclonaux y compris avec l'association casirivimab et imdevimab pendant et au décours de l'administration. Si des signes ou des symptômes cliniquement significatifs d'une réaction d'hypersensibilité ou d'anaphylaxie apparaissent, arrêtez immédiatement la perfusion et débutez un traitement approprié. Aucune autre administration ne devra être réalisée.
Des réactions liées à la perfusion ont été observées avec l'administration de l'association casirivimab et imdevimab. Ces réactions peuvent être sévères ou mettre en jeu le pronostic vital. Les signes et symptômes des réactions liées à la perfusion peuvent inclure sans s'y limiter : fièvre, frissons, nausées, céphalées, bronchospasme, diminution de la saturation en oxygène, difficulté respiratoire, hypotension, hypertension, arythmie (par exemple fibrillation auriculaire, tachycardie, bradycardie), douleur ou inconfort thoracique, angio-oedème, irritation de la gorge, fatigue, altération de l'état mental, éruption cutanée y compris urticaire, prurit, myalgie, vertiges, diaphorèse. Si des signes ou symptômes apparaissent pendant l'administration, il convient de ralentir le débit de perfusion ou d'arrêter la perfusion et de débuter un traitement approprié.
Aussi, il convient de toujours disposer d'un traitement médical approprié et de surveiller de manière rapprochée les patients pendant toute la durée de la perfusion et pendant au moins une heure après l'arrêt du traitement.
Surveillance virologique
· Traitement
Les mutations en position 484 de la protéine de spicule virale (e.g E484K/Q) sont péjoratives pour l'activité du casirivimab alors que l'association casirivimab et imdevimab conserve une activité neutralisante. Cependant, un risque d'émergence de mutations sous traitement n'est pas exclu.
Un test de criblage pour la détection d'éventuelles mutations pouvant impacter l'efficacité du casirivimab, notamment la mutation en position 484 (ou d'autres mutations selon les préconisations des CNR des virus respiratoires), devra être systématiquement demandé avant le traitement pour adapter la surveillance virologique si nécessaire.
Le traitement par casirivimab et imdevimab pourra être administré sans attendre le résultat du test de criblage, y compris dans les territoires où la circulation des variants porteurs d'une mutation en position 484 (ou d'autres mutations selon les préconisations des CNR des virus respiratoires) est supérieure à 10%.
Si le traitement a été administré avant le résultat du test de criblage et qu'il s'avère que le patient est infecté par un variant porteur d'une mutation en position 484 (ou d'autres mutations selon les préconisations des CNR des virus respiratoires), il conviendra de renforcer le suivi virologique en collégialité entre cliniciens et virologues et de mettre en place des mesures d'isolement tenant compte des recommandations en vigueur.
Tous les patients feront l'objet d'une surveillance virologique à J7 post-traitement incluant une détection moléculaire du génome viral et une recherche de mutations pouvant être péjoratives pour la réponse au casirivimab notamment en position 484 de la spicule virale (ou d'autres mutations selon les préconisations des CNR des virus respiratoires). En fonction des résultats, le calendrier des visites supplémentaires sera adapté en collégialité entre cliniciens et virologues avec des mesures d'isolement à mettre en place en tenant compte des recommandations en vigueur.
· Prophylaxie post-exposition
Un test virologique de détection du SARS-CoV-2 négatif est nécessaire avant la mise en place d'un traitement en prophylaxie post-exposition.
Tous les patients feront l'objet d'une surveillance virologique à J7 post-traitement par un test RT-PCR sur prélèvement nasopharyngé, et s'il s'avérait positif, une détection moléculaire du génome viral et une recherche de mutations pouvant être péjoratives pour la réponse au casirivimab notamment en position 484 de la spicule virale (ou d'autres mutations selon les préconisations des CNR des virus respiratoires). En fonction des résultats, le calendrier des visites supplémentaires sera adapté en collégialité entre cliniciens et virologues avec des mesures d'isolement à mettre en place en tenant compte des recommandations en vigueur.
· Prophylaxie pré-exposition
Un test virologique de détection du SARS-CoV-2 négatif est nécessaire avant la mise en place d'un traitement en prophylaxie pré-exposition.
Tous les patients feront l'objet d'une surveillance virologique et immunologique avant chaque administration du casirivimab et de l'imdevimab par un test RT-PCR sur prélèvement nasopharyngé et un dosage des anticorps anti-S et anti-N. Si le test RT-PCR s'avérait positif, une détection moléculaire du génome viral et une recherche de mutations pouvant être péjoratives pour la réponse au casirivimab notamment en position 484 de la spicule virale (ou d'autres mutations selon les préconisations des CNR des virus respiratoires) devront être réalisées. Le traitement prophylactique devra être arrêté et la prise en charge du patient réévaluée.
En fonction des résultats des tests virologiques, le calendrier des visites supplémentaires sera adapté en collégialité entre cliniciens et virologues avec des mesures d'isolement à mettre en place en tenant compte des recommandations en vigueur.
Formes sévères de la COVID 19
Un risque d'aggravation de l'état clinique du patient ne peut être exclu avec les anticorps monoclonaux, tels que le casirivimab et l'imdevimab, lorsqu'ils sont administrés à des patients hospitalisés du fait de la COVID-19 et nécessitant de l'oxygène à haut débit ou une ventilation mécanique.
L'association casirivimab et imdevimab ne doit pas être utilisée chez les patients SARS- CoV2 positifs nécessitant une oxygénothérapie invasive du fait de la COVID-19, le bénéfice de l'association n'ayant pas été démontré pour ces patients.
Résumé du profil de tolérance
Au total, environ 7116 individus (environ 4666 par voie IV et 2450 par voie SC) ont été exposés à l'association casirivimab et imdevimab dans le cadre des essais cliniques soutenant les indications mentionnées. Le profil de sécurité de l'administration IV est principalement basé sur l'analyse des données de sécurité combinées des phases 1/2/3 de l'étude COV 2067 tandis que pour la voie sous-cutanée, le profil de sécurité est principalement basé sur l'étude COV-2069. Une analyse élargie a également été réalisée sur les données de sécurité d'études complémentaires (COV-20145, HV-2093) (voir rubrique Propriétés pharmacodynamiques).
Les effets indésirables signalés dans le cadre du programme de développement clinique concernent des réactions d'hypersensibilité, notamment des réactions liées à la perfusion et des réactions au site d'injection.
Tableau récapitulatif des effets indésirables
Tableau 1 : Les effets indésirables sont classés par classes systèmes organes en appliquant les conventions suivantes : très fréquent (≥ 1/10), fréquent ≥ 1/100, < 1/10), peu fréquent (≥ 1/1000, < 1/100), rare (≥ 1/10 000, < 1/1000), très rare (< 1/10 000), fréquence indéterminée (ne peut être estimée sur la base des données disponibles). Au sein de chaque groupe de fréquence, les effets indésirables sont présentés suivant un ordre décroissant de gravité.
Tableau 1: Liste des effets indésirables survenus chez les patients traités par l'association du casirivimab et de l'imdevimab dans le cadre des essais cliniques :
| Système organe-classe | Effets indésirable | Catégorie de fréquence |
| administration IV | ||
| Troubles du système immunitaire | Anaphylaxie3, hypersensibilité | Très rare |
| Affections du système nerveux | Vertiges2* | Peu fréquent |
| Affections vasculaires | Bouffées vasomotrices2* | Rare |
| Affections gastro-intestinales | Nausées2* | Peu fréquent |
| Affections de la peau et du tissu sous-cutané | Eruption cutanée2* | Peu fréquent |
| Urticaire2* | Rare | |
| Troubles généraux et anomalies au site d'administration | Frissons2* | Peu fréquent |
| Lésions, intoxications et complications liées aux procédures | Réactions liées à la perfusion2 | Peu fréquent |
| administration SC | ||
| Affections hématologiques et du système lymphatique | Lymphadénopathie 1,4* | Peu fréquent |
| Affections du système nerveux | Vertiges5* | Peu fréquent |
| Affections de la peau et du tissu sous-cutané | Prurit5* | Rare |
| Troubles généraux et anomalies au site d'administration | Réactions au site d'injection5 | Fréquent |
| 1 Observé dans l'étude HV-2093 avec administration SC répétée 2 Fréquence déterminée à partir de l'étude COV 2067 3 Fréquence déterminée en se basant sur toutes les études, c'est-à-dire à la fois IV et SC. (2066, 2067, 2069, 20145 and 2093) 4 Fréquence déterminée à partir de l'étude HV 2093 (étude avec doses répétées SC) 5 Fréquence déterminée à partir de l'étude COV 2069 * Dans certains cas, les symptômes des réactions liées à la perfusion et des réactions au site d'injection ont été signalés comme des effets indésirables individuels | ||
Les données préliminaires de
sécurité d'emploi issues de l'étude Recovery ne mettent pas en évidence
de nouveau signal de pharmacovigilance.
Description de certains effets indésirables
Hypersensibilité, y compris anaphylaxie et réactions liées à la perfusion :
Des réactions d'hypersensibilité de sévérité variable ont été observées au cours du programme de développement clinique.
Une réaction anaphylactique a été observée dans le programme de développement clinique. L'effet a commencé dans l'heure suivant la fin de la perfusion et a nécessité un traitement comprenant de l'épinéphrine. (voir rubrique Mises en garde spéciales et précautions d'emploi).
Réactions liées à la perfusion:
Des réactions liées à la perfusion ont été observées avec l'administration IV de casirivimab et d'imdevimab dans tous les groupes posologiques dans le cadre des études cliniques. Ces réactions étaient pour la plupart d'intensité légère à modérée et ont été généralement observées pendant ou dans les 24 heures suivant la perfusion et se sont résolues soit sans intervention, soit avec les soins habituels. Les signes et symptômes fréquemment rapportés pour les réactions liées à la perfusion comprenaient des nausées, des frissons, des étourdissements (ou syncope), des éruptions cutanées, de l'urticaire et des bouffées vasomotrices. D'autres présentations cliniques connues des réactions liées à la perfusion peuvent également être attendues (voir rubrique Mises en garde spéciales et précautions d'emploi).
Réactions au site d'injection :
Des réactions au site d'injection ont été rapportées dans toutes les études avec l'administration sous-cutanée, qu'il s'agisse d'une dose unique ou de doses répétées. Ces réactions étaient en majorité locales, d'intensité légère à modérée et se sont résolues sans traitement soit avec une prise en charge selon les pratiques courantes. Les signes et symptômes les plus fréquemment rapportés pour ces réactions étaient érythème, prurit, ecchymose, oedème, douleur/sensibilité et urticaire. Dans l'étude à doses répétées HV-2093 des lymphadénopathies localisées ont également été observées. Neuf participants ayant reçu le casirivimab et l'imdevimab ont arrêté le traitement prophylactique en raison d'un évènement indésirable dont deux en raison d'une infection par le SARS-CoV-2.
Population pédiatrique :
Administration IV (Population traitée) : Aucune donnée n'est disponible pour les patients pédiatriques de moins de 18 ans.
Administration sous-cutanée : 45 (3 %) et 21 (14 %) adolescents âgés de ≥ 12 et < 18 ans ont reçu un traitement par casirivimab et imdevimab dans l'étude COV-2069, respectivement dans les cohortes A et B, et le profil de sécurité observé était similaire à celui des patients adultes.
Population âgée :
Administration IV : Dans les études COV-2067, 485 (12 %) patients âgés de ≥ 65 ans ont reçu un traitement par casirivimab et imdevimab. Le profil de sécurité de ces patients était similaire à celui des patients adultes de moins de 65 ans.
Administration sous-cutanée : Dans les études COV-2069 (cohortes A et B) et HV-2093, un total de 120 (9 %, cohorte A), 15 (10,0 %, cohorte B) et 90 (12 %, HV-2093) individus âgés de ≥ 65 ans ont été traités par casirivimab et imdevimab et le profil d'innocuité était similaire à celui des adultes de moins de 65 ans.
Déclaration des effets indésirables suspectés
La déclaration des effets indésirables suspectés est importante. Elle permet une surveillance continue du rapport bénéfice / risque du médicament. Les professionnels de santé déclarent tout effet indésirable suspecté via le système national de déclaration : Agence nationale de sécurité du médicament et des produits de santé (ANSM) et réseau des Centres Régionaux de Pharmacovigilance - Site internet : signalement.social-sante.gouv.fr.
AVANT LE TRAITEMENT réaliser un test de criblage pour la détection d'éventuels variants porteurs de la mutation E484K (ou d'autres variants selon les éventuelles préconisations des CNR des virus respiratoires) pour adapter la surveillance virologique.
Le RESULTAT de ce test
est OBLIGATOIRE pour l'administration du traitement dans les territoires où la
circulation de ces variants est > 10% .
SURVEILLANCE clinique des patients pendant l'administration et les OBSERVER pendant au moins une heure après complète administration de la perfusion.
Grossesse
Il existe peu de données sur l'utilisation du casirivimab et de l'imdevimab chez les femmes enceintes. Aucune étude de toxicité sur la reproduction animale n'a été réalisée, cependant, une étude de réactivité tissulaire croisée en utilisant des tissus humains foetaux et les anticorps casirivimab et imdevimab n'a pas décelé de fixation préoccupante sur le plan clinique (voir rubrique Données de sécurité préclinique). Il est décrit que les anticorps humains d'isotype IgG1 traversent la barrière placentaire, par conséquent, le casirivimab et l'imdevimab peuvent être transférés de la mère au foetus lors de la grossesse. A ce jour, l'impact de ce transfert potentiel du casirivimab et de l'imdevimab pour le foetus n'est pas connu.
Le casirivimab et l'imdevimab ne doivent pas être utilisés pendant la grossesse sauf si le bénéfice potentiel justifie le risque potentiel pour la mère et le foetus.
Allaitement
Il n'y a pas de données disponibles sur l'excrétion du casirivimab ou de l'imdevimab dans le lait maternel. Un risque pour les nouveau-nés/nourrissons ne peut être exclu.
Les IgG maternelles sont présentes dans le lait maternel. Les bénéfices de l'allaitement pour le développement et la santé du nourrisson doivent être pris en compte en même temps que les besoins cliniques de la mère d'être traitée par le casirivimab et l'imdevimab mais également en évaluant le risque potentiel de tout effet indésirable du casirivimab et de l'imdevimab sur l'enfant allaité ou de l'infection maternelle sous-jacente.
Les patientes atteintes de la COVID-19 qui allaitent doivent appliquer les précautions nécessaires selon les recommandations en vigueur pour éviter d'exposer le nourrisson au virus.
Fertilité
Il n'y a pas eu d'étude permettant d'évaluer les effets du casirivimab et de l'imdevimab sur la reproduction.
Aucune étude d'interaction n'a été réalisée. Le casirivimab et l'imdevimab sont des anticorps monoclonaux qui ne sont pas excrétés par voie rénale ni métabolisés par les enzymes du cytochrome P450 ; il est donc peu probable qu'il y ait des interactions avec la prise concomitante de médicaments qui sont excrétés par voie rénale ou qui sont des substrats, des inducteurs ou des inhibiteurs des enzymes du cytochrome P450.
Le casirivimab et l'imdevimab se lient aux épitopes de la protéine de spicule du SARS-CoV-2 qui ont été utilisés comme protéine immunogène dans tous les vaccins développés contre la COVID-19. Il est donc possible que le casirivimab et l'imdevimab puissent avoir un impact sur les réponses aux vaccins contre la COVID-19.
Il est recommandé d'appliquer un délai minimum de 3 mois entre la dernière administration de casirivimab et imdevimab et la réalisation d'une vaccination contre la COVID-19.
Le traitement par l'association casirivimab et imdevimab doit être administré et supervisé par un professionnel de santé qualifié dans un établissement de santé. Le traitement doit être administré dans des conditions permettant de prendre en charge une réaction liée à la perfusion ou une réaction allergique.
Administration intraveineuse (IV)
Casirivimab et imdevimab doivent être administrés ensemble dans une même perfusion IV.
Administration sous-cutanée (SC)
Casirivimab et imdevimab doivent être administrés de manière consécutive par injection SC.
Durée et surveillance du traitement
4 Adultes ou enfants de 12 ans et plus
Les patients sont surveillés cliniquement pendant l'administration et observés pendant au moins une heure après l'administration.
Posologie
Traitement
Le traitement doit être initié le plus rapidement possible après l'obtention du test virologique positif.
Voir la rubrique Mises en garde spéciales et précautions d'emploi concernant la réalisation d'un test de criblage pour adapter la surveillance virologique.
Dose recommandée
· Patient ne nécessitant pas d'oxygénothérapie du fait de la COVID-19 : le traitement doit être instauré dès que possible après l'obtention du test RT-PCR au SARS-CoV-2 positif et dans un délai maximum de 5 jours :
- La dose recommandée de l'association casirivimab et imdevimab est une dose unique de 600 mg de casirivimab et 600 mg d'imdevimab administrés en une seule perfusion intraveineuse.
· Patient hospitalisé du fait de la COVID-19 et séronégatifs (IgG anti-S) nécessitant une oxygénothérapie non invasive :
- La dose recommandée de l'association casirivimab et imdevimab est une dose unique de 4000 mg de casirivimab et 4000 mg d'imdevimab administrés en une seule perfusion intraveineuse de 250mL pendant une durée de 60mn (+/-15mn).
Casirivimab et imdevimab doivent être administrés ensemble dans une même perfusion intraveineuse.
Voir la rubrique Précautions particulières d'élimination et de manipulation pour les recommandations de dilution et d'administration.
Prophylaxie post-exposition
La dose recommandée de l'association casirivimab et imdevimab est une dose unique de 600 mg de casirivimab et 600 mg d'imdevimab administrés ensemble en une seule perfusion IV à l'aide d'une pompe ou par gravité (voir tableau 16).
En cas d'impossibilité d'utiliser la voie IV, le casirivimab et l'imdevimab peuvent être administrés de manière consécutive par injection SC (voir tableau 19).
Le casirivimab et l'imdevimab doivent être administrés dès que possible après une exposition confirmée au SARS-CoV-2.
Prophylaxie pré-exposition
La dose recommandée de l'association casirivimab et imdevimab est une dose initiale de 600 mg de casirivimab et 600 mg d'imdevimab administrés ensemble soit en perfusion IV unique à l'aide d'une pompe ou par gravité (voir tableau 16), soit par injections SC consécutives (voir tableau 19).
En cas d'exposition continue ou prolongée au SARS-CoV-2, il est nécessaire de réitérer l'administration de l'association casirivimab et imdevimab toutes les 4 semaines selon le schéma suivant :
● Une dose initiale de 600 mg de casirivimab et 600 mg d'imdevimab par perfusion IV ou injection SC.
● Puis, toutes les 4 semaines, une dose de 300 mg de casirivimab et 300 mg d'imdevimab par perfusion IV ou injection SC.
● Il est possible de changer le mode d'administration IV ou SC au cours du traitement.
Omission d'une dose
Si une dose de l'association casirivimab et imdevimab est oubliée, elle doit être administrée dès que possible. Le calendrier d'administration doit être ajusté afin de maintenir l'intervalle de quatre semaines entre les doses.
Populations particulières
Insuffisance rénale
Aucune adaptation posologique n'est recommandée chez les patients atteints d'insuffisance rénale (voir rubrique Propriétés pharmacocinétiques).
Insuffisance hépatique
La pharmacocinétique de l'association de casirivimab et imdevimab n'a pas été étudiée chez les patients atteints d'insuffisance hépatique. La nécessité d'une adaptation de la posologie chez les patients atteints d'insuffisance hépatique n'est pas établie (voir rubrique Propriétés pharmacocinétiques).
Population pédiatrique
La sécurité et l'efficacité de l'association casirivimab et imdevimab chez les enfants de moins de 12 ans n'ont pas encore été établies. Aucune donnée disponible.
Aucun ajustement posologique n'est recommandé chez les enfants âgés de 12 ans et plus et pesant au moins 40 kg.
Mode d'administration
Casirivimab et imdevimab sont destinés à être administrés en association par perfusion IV ou injection SC. Ne pas les administrer par injection intramusculaire (IM).
Pour les instructions concernant les modalités de dilution et d'administration de l'association casirivimab et imdevimab, voir la rubrique Précautions particulières d'élimination et de manipulation.
Lors d'une administration par voie IV, casirivimab et imdevimab doivent être administrés en association au moyen d'une ligne de perfusion intraveineuse munie d'un filtre stérile d'appoint ou en ligne de 0,2 micron.
Durée de conservation :
Flacon fermé
24 mois
Flacons unidoses de 6 ml
Le médicament doit être utilisé immédiatement, tout produit restant doit être jeté.
Solution diluée pour une administration par voie intraveineuse
La solution en flacon doit être diluée avant l'administration. La solution pour perfusion qui a été préparée est destinée à être utilisée immédiatement. La stabilité physico-chimique en cours d'utilisation a été démontrée pendant 36 heures à 2-8°C ou 4 heures à une température ne dépassant pas 25°C. D'un point de vue microbiologique, le produit doit être utilisé immédiatement. En cas d'utilisation non immédiate, les durées et conditions de conservation en cours d'utilisation relèvent de la responsabilité de l'utilisateur et ne devraient normalement pas dépasser 24 heures à 2-8°C, sauf si la méthode de dilution prévient tout risque de contamination microbienne.
La solution pour perfusion qui a été préparée est réfrigérée, laissez la poche de perfusion IV s'équilibrer à température ambiante pendant environ 30 minutes avant l'administration.
Conservation des seringues pour administration sous-cutanée
Ce produit est sans conservateur et par conséquent, les seringues préparées doivent être administrées immédiatement. La stabilité physico-chimique en cours d'utilisation a été démontrée pendant 36 heures à 2-8°C ou 4 heures à une température ne dépassant pas 25°C. D'un point de vue microbiologique, le produit doit être utilisé immédiatement. En cas d'utilisation non immédiate, les durées et conditions de conservation en cours d'utilisation relèvent de la responsabilité de l'utilisateur et ne devraient normalement pas dépasser 24 heures à 2-8°C, sauf si la méthode de dilution prévient tout risque de contamination microbienne. Si les seringues sont réfrigérées, laissez-les s'équilibrer à température ambiante pendant environ 10 à 15 minutes avant l'administration.
Précautions particulières de conservation :
A conserver au réfrigérateur (entre 2°C et 8°C) dans l'emballage d'origine à l'abri de la lumière.
Ne pas congeler. Ne pas agiter.
Pour les conditions de conservation du médicament après ouverture et dilution, voir rubrique Durée de conservation.
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
Il n'y a pas d'expérience connue chez l'Homme de surdosage aigu avec l'association du casirivimab et de l'imdevimab. Des doses allant jusqu'à 8000 mg (4000 mg de casirivimab et 4000 mg d'imdevimab) ont été administrées dans les essais cliniques sans toxicité limitant la dose. Le traitement d'un surdosage doit comprendre des mesures générales de soutien, y compris la surveillance des signes vitaux et l'observation de l'état clinique du patient. Il n'y a pas d'antidote spécifique en cas de surdosage avec l'association de casirivimab et imdevimab.
Mécanisme d'action
Tableau 2: Données
de neutralisation sur des virus pseudotypés pour la séquence complète
ou des substitutions de la protéine de spicule du SARS-CoV-2 portées
par les variants “préoccupants” ou “à suivre” avec le casirivimab et l'imdevimab seuls ou en association.
Le casirivimab et l'imdevimab est une association de deux anticorps monoclonaux recombinants de type IgG1 non modifiés au niveau des régions du fragment constant (Fc), où chaque anticorps cible la protéine de spicule (S) du SARS-CoV-2. Le casirivimab et l'imdevimab ciblent sur la protéine S des épitopes différents et non chevauchants qui sont situés dans le domaine de liaison avec son récepteur cible. Le blocage de l'interaction de la protéine S avec l'enzyme de conversion de l'angiotensine 2 (ACE2) empêche le SARS-CoV- 2 de se lier au récepteur ACE2, prévenant ainsi l'entrée du virus dans les cellules humaines ce qui permet une inhibition de l'infection des cellules hôtes.
Activité antivirale
L'activité neutralisante in vitro du casirivimab, de l'imdevimab et du casirivimab en association avec l'imdevimab contre le SARS-CoV-2 a été mesurée en détectant la neutralisation du virus dans un modèle dose-réponse utilisant une culture de cellules Vero E6. Il a été montré lors des tests de séroneutralisation par réduction des plages de lyse (PRNT50) que le casirivimab, l'imdevimab et le casirivimab en association avec l'imdevimab ont permis une inhibition de 50 % de l'infection du SARS-CoV-2 (isolat USA-WA1 / 2020) à une concentration respective de 37,4 pM (0,006 µg / mL), 42,1 pM (0,006 µg / mL) et 31,0 pM (0,005 µg / mL).
Risque d'ADE (Antibody Dependent Enhancement) de l'infection
Le potentiel du casirivimab et de l'imdevimab pour médier l'entrée virale dans les cellules a été évalué dans des lignées cellulaires immunitaires avec des pseudoparticules du virus de la stomatite vésiculeuse (VSV) recombinant exprimant la protéine de spicule du SARS-CoV- 2 en présence du casirivimab et/ou d'imdevimab à des concentrations jusqu'à environ 10 fois inférieures à leur pouvoir de neutralisation (EC50). L'association du casirivimab et de l'imdevimab ainsi que l'imdevimab seul, mais pas le casirivimab seul, ont médié l'entrée de pseudoparticules dans les cellules FcγR2+ Raji (infection maximum des cellules de 0,69% pour l'association des deux anticorps et 1,34% pour l'imdevimab seul) et FcγR1+/FcγR2+ THP1 (infection maximum des cellules de 0,06 % pour l'association des deux anticorps et 0,24 % pour l'imdevimab seul). Aucune infection n'a été observée dans les autres lignées cellulaires testées (cellules IM9, K562, Ramos et U937).
L'effet in vivo du casirivimab et de l'imdevimab a été évalué chez des macaques rhésus et des hamsters dorés syriens.
Résistance antivirale
Il existe un risque potentiel d'échec du traitement dû à l'émergence de variants du virus SARS-CoV-2 résistants à l'association casirivimab et imdevimab. Les médecins prescripteurs doivent tenir compte, lorsque ces données sont disponibles, de la prévalence de la circulation des variants du SARS-CoV-2 dans leur territoire lorsqu'ils envisagent l'administration de l'association casirivimab et imdevimab.
L'association de ces deux anticorps ciblant différents épitopes est susceptible de limiter le risque d'émergence de variants résistants sous traitement.
Les données disponibles, qui sont susceptibles d'évoluer en lien avec les préconisations des CNR des virus respiratoires, suggèrent un maintien de l'activité de chacun des composants de l'association sur le variant alpha 20I/501Y.V1, lignée B.1.1.7 (sans la mutation E484K) et le variant delta (lignée B.1.617.2)
Selon les données in vitro de neutralisation, la mutation E484K commune aux variants bêta (20H/501Y.V2, lignée B.1.351) et gamma (20J/501Y.V3, lignée P.1 ou B.1.1.248) est péjorative pour l'activité d'un des deux composants de l'association, le casirivimab.
Données in vitro :
Le casirivimab et l'imdevimab sont deux anticorps qui se lient à des épitopes distincts et non chevauchants de la protéine de spicule du SARS-CoV-2, réduisant ainsi la probabilité de résistance virale. Le pouvoir de neutralisation du casirivimab, de l'imdevimab et de l'association des deux anticorps a été évalué par rapport aux différents variants de la protéine de spicule, y compris les variants dits “préoccupants” (variants of concern), les “variants à suivre” (variants of interest), les variants identifiées dans les études d'échappement in vitro et les variations génétiques du SARS-CoV-2 qui sont surveillées au niveau mondial et qui sont partagées dans des bases de données internationales, dont la base de données GISAID (Global Initiative on Sharing Avian Influenza Data)
Dans les essais de pseudo-neutralisation in vitro, l'association du casirivimab et de l'imdevimab conserve son pouvoir neutralisant contre tous les variants testés (sous forme de pseudoparticules virales ou de variants authentiques), tels qu'indiqué dans les tableaux 2 et 3, même si le casirivimab seul présente une diminution de son efficacité contre certains variants. Par conséquent, l'association du casirivimab et de l'imdevimab devrait maintenir une activité de neutralisation contre les variants “préoccupants” et à “suivre”.
| Lignée avec des substitutions de la protéine de spicule | Substitutions clés testées | Susceptibilité réduite à l'association du casirivimab et de l'imdevimab | Susceptibilité réduite au casirivimab | Susceptibilité réduite à l'imdevimab |
| B.1.1.7 (UK origin/Alpha) | Protéine de spicule entièrea | pas de changementd | pas de changementd | pas de changementd |
| B.1.351 (South Africa origin/Beta) | Protéine de spicule entièreb | pas de changementd | 45-fois | pas de changementd |
| P.1 (Brazil origin/Gamma) | Protéine de spicule entièrec | pas de changementd | 418-fois | pas de changementd |
| B.1.427/B.1.429 (California origin/Epsilon) | L452R | pas de changementd | pas de changementd | pas de changementd |
| B.1.526 (New York origin/Iota)e | E484K | pas de changementd | 25-fois | pas de changementd |
| B.1.617.1/B.1.617.3 (India origin/Kappa) | L452R+E484Q | pas de changementd | 7-fois | pas de changementd |
| B.1.617.2 (India origin/Delta) | L452R+T478K | pas de changementd | pas de changementd | pas de changementd |
a Pseudovirus exprimant la totalité de la protéine de spicule du variant a été testé. Liste des substitutions par rapport à la protéine de spicule de type sauvage: del69-70, del145, N501Y, A570D, D614G, P681H, T716I, S982A, D1118H.
b Pseudovirus exprimant la totalité de la protéine de spicule du variant a été testé. Liste des substitutions par rapport à la protéine de spicule de type sauvage: D80Y, D215Y, del241-243, K417N, E484K, N501Y, D614G, A701V.
c Pseudovirus exprimant la totalité de la protéine de spicule du variants a été testé. Liste des substitutions par rapport à la protéine de spicule de type sauvage: L18F, T20N, P26S, D138Y, R190S, K417T, E484K, N501Y, D614G, H655Y, T1027I, V1176F
d Pas de changement: Réduction de la sensibilité ≤ 5 fois.
e Tous les isolats de la lignée New York ne portent pas la substitution E484K (en février 2021).
Tableau 3: Données de neutralisation sur les variants authentiques du SARS-CoV-2 avec le casirivimab et l'imdevimab seuls ou en association
| Lignée avec substitution de la protéine de spicule | Susceptibilité réduite à l'association du casirivimab et de l'imdevimab | Susceptibilité réduite au casirivimab | Susceptibilité réduite à l'imdevimab |
| B.1.1.7 (UK origin/alpha) | pas de changementa | pas de changementa | pas de changementa |
| B.1.351 (South Africa origin/beta) | pas de changementa | 5-fois | pas de changementa |
| B.1.617.1 (India origin/Kappa) | pas de changementa | 6-fois | pas de changementa |
a Pas de changement: Réduction de la sensibilité ≤ 5 fois.
Le Tableau 4 présente une liste complète des substitutions de la protéine de spicule du SARS-CoV-2 avec une sensibilité réduite ≥ 5 fois au casirivimab ou à l'imdevimab seuls, ou à l'association des deux anticorps
Tableau 4: Données de neutralisation sur des pseudovirus pour différentes substitutions du SARS-CoV-2 qui ont un impact sur le pouvoir de neutralisation du casirivimab et de l'imdevimab seuls ou en association
| Substitution du SARS-CoV-2 | Susceptibilité réduite à l'association du casirivimab et de l'imdevimab | Susceptibilité réduite au casirivimab | Susceptibilité réduite à l'imdevimab |
| K417E | Pas de changementa | 182-fois | Pas de changementa |
| K417N | Pas de changementa | 7-fois | Pas de changementa |
| K417R | Pas de changementa | 61-fois | Pas de changementa |
| Y453F | Pas de changementa | > 438-fois | Pas de changementa |
| L455F | Pas de changementa | 80-fois | Pas de changementa |
| E484K | Pas de changementa | 25-fois | Pas de changementa |
| F486V | Pas de changementa | > 438-fois | Pas de changementa |
| Q493K | Pas de changementa | > 438-fois | Pas de changementa |
| K444N | Pas de changementa | Pas de changementa | > 755-fois |
| K444Q | Pas de changementa | Pas de changementa | > 548-fois |
| K444T | 6-fois | Pas de changementa | > 1033-fois |
| V445A | Pas de changementa | Pas de changementa | 548-fois |
| V445T | Pas de changementa | 107-fois | Pas de changementa |
| E406D | Pas de changementa | 51-fois | Pas de changementa |
| G485D | Pas de changementa | 5-fois | Pas de changementa |
| G476S | Pas de changementa | 5-fois | Pas de changementa |
| F486L | Pas de changementa | 61-fois | Pas de changementa |
| F486S | Pas de changementa | > 715-fois | Pas de changementa |
| P337L | Pas de changementa | Pas de changementa | 5-fois |
| N439K | Pas de changementa | Pas de changementa | 463-fois |
| N440K | Pas de changementa | Pas de changementa | 28-fois |
| K444L | Pas de changementa | Pas de changementa | 153-fois |
| K444M | Pas de changementa | Pas de changementa | 1577-fois |
| G446V | Pas de changementa | Pas de changementa | 135-fois |
| N450D | Pas de changementa | Pas de changementa | 9-fois |
| Q498H | Pas de changementa | Pas de changementa | 17-fois |
| P499S | Pas de changementa | Pas de changementa | 206-fois |
| E484Q | Pas de changementa | 19-fois | Pas de changementa |
| Q493E | Pas de changementa | 446-fois | Pas de changementa |
| G476D | Pas de changementa | 1021 fois | Pas de changementa |
a Pasdechangement: Réduction de la sensibilité ≤ 5 fois.
Le tableau 5 présente une liste complète des substitutions avec une sensibilité réduite < 5-fois au casirivimab ou l'imdevimab seuls et à l'association des deux anticorps.
Tableau 5: Données de neutralisation sur des pseudovirus pour différentes substitutions du SARS-CoV-2 qui n'ont pas d'impact sur le pouvoir de neutralisation du casirivimab ou de l'imdevimab seul ou en association
| Substitution du SARS-CoV-2 | |||||
| L18F | R346E | V382L | K458N | G485D | H519Q |
| W152C | R346G | P384L | K458Ra | G485S | A520S |
| A222V | R346K | P384S | I468V | F490L | A522S |
| Q321La | A348Ta | R403K | T470I | F490P | A522V |
| P322A | A352S | R408Ia | E471Q | F490S | K537R |
| T323I | N354Da | Q409Ea | I472Va | F490Y | D614G |
| P330S | N354S | Q414E | A475V | S494P | D614N |
| E340A | S359Na | Q414R | S477N | N501Y | V687G |
| E340D | V367Fa | A435Sa | T478I | G504D | V1128A |
| E340K | N370S | N439V | T478K | G504S | |
| V341Ia | A372T | L441Q | P479S | Y508Ha | |
| A344Sa | F377L | Y449N | V483Aa | E516Q | |
| T345P | K378Ra | L452R | V483F | H519Pa | |
a Non évaluépour le casirivimab et l'imdevimab en association.
La corrélation entre les données in vitro sur les pseudovirus et les conséquences en clinique n'est pas connue à ce jour.
Atténuation de la réponse immunitaire
L'administration d'anticorps comporte, en théorie, un risque d'atténuation de la réponse immunitaire endogène face au SARS-CoV-2 et une augmentation de la vulnérabilité des patients à une réinfection.
Effets pharmacodynamiques
Etude COV-2067:
L'essai COV-2067 a évalué l'association du casirivimab et de l'imdevimab avec des doses jusqu'à 7 fois la dose recommandée (doses allant de 600 mg à 4000mg de casirivimab ou d'imdevimab) chez des patients ambulatoires atteints de la COVID-19. Aucune relation dose- réponse pour l'efficacité n'a été identifiée pour l'association des deux anticorps à toutes les doses, sur la base de la charge virale et des résultats cliniques. Des réductions similaires de la charge virale ont été observées chez les participants pour les doses IV (600 mg de casirivimab et 600 mg d'imdevimab) et sous-cutanées (600 mg de casirivimab et 600 mg d'imdevimab).
Etude COV-2045 :
COV-2045 est une étude de phase 2 randomisée, en double aveugle, contrôlée par placebo, en groupes parallèles pour évaluer le profil dose-réponse de doses uniques IV ou sous- cutanées de l'association casirivimab et imdevimab chez des patients ambulatoires (symptomatiques sans facteur de risque ou asymptomatiques) infectés par le SARS-CoV-2. Le traitement a été initié dans les 3 jours suivant l'obtention d'un résultat positif au test d'infection par le SARS-CoV-2 chez 803 patients, parmi lesquels 116 participants ont reçus une dose IV de 1 200 mg de l'association casirivimab et imdevimab (600 mg de casirivimab et 600 mg d'imdevimab).
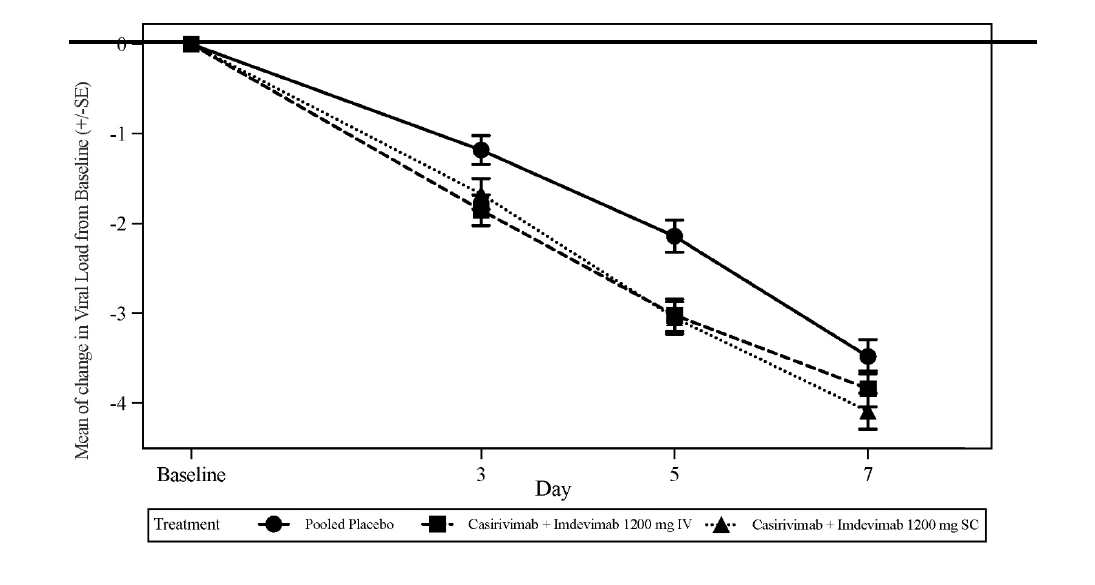
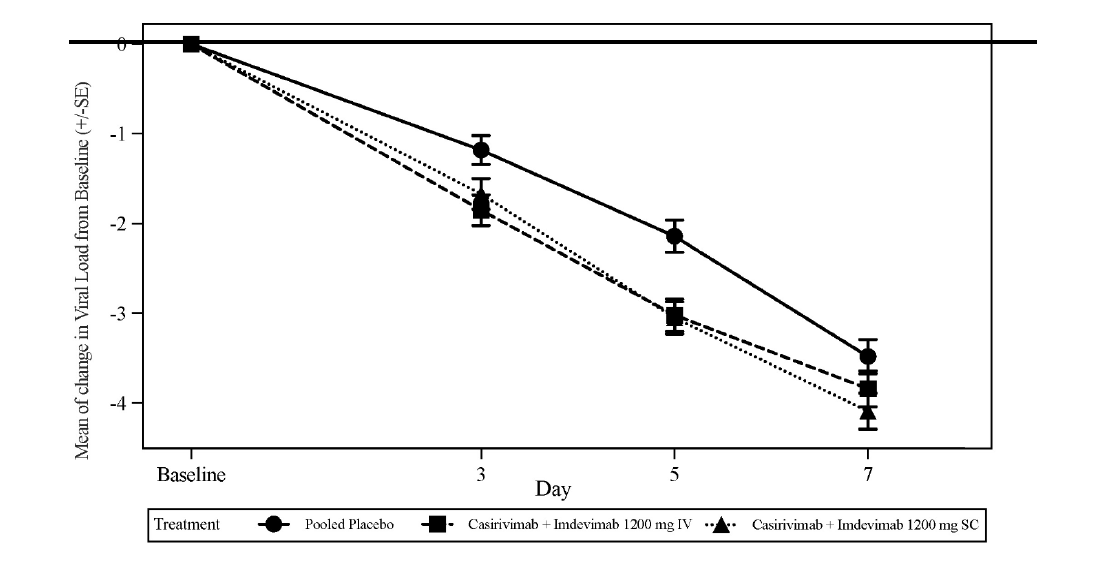
Le critère d'évaluation principal pré-spécifié était la variation quotidienne moyenne pondérée dans le temps (TWA) de la charge virale (log10 copies/mL) par rapport à l'inclusion, qui a été mesurée par RT-qPCR à partir d'échantillons d'écouvillonnage nasopharyngé, du jour 1 au jour 7 (chez les patients avec un résultat de RT-qPCR positif au SARS-CoV-2 et séronégatif à l'inclusion, définie comme la population mFAS séronégative). Le traitement par 1 200 mg IV de l'association des deux anticorps a entraîné une réduction statistiquement significative de la TWA entre la valeur initiale et le jour 7 de la charge virale par rapport au placebo (-0,56 log10 copies/mL, p < 0,0007). Les réductions les plus importantes de la charge virale par rapport au placebo se sont produites chez les patients ayant une charge virale élevée (> 107 copies/ml) avec une différence de TWA du jour 1 au jour 7 de -0,85 log10 copies/ml (p < 0,0001). La figure 1 représente la variation moyenne par rapport à l'inclusion de la charge virale du SARS-CoV-2 au cours du temps.
Figure 1: Variation moyenne de la charge virale (log10 copies/mL) à chaque visite de l'inclusion au jour 7 chez les patients recevant 1 200 mg IV et 1 200 mg SC (séronégatif mFAS) Étude COV-20145
Données d'efficacité
L'ATU de cohorte et l'accès précoce sont octroyés pour une utilisation à titre exceptionnel au cas par cas au regard du contexte sanitaire lié à la pandémie de COVID-19, des données de mortalité dans la population cible, et de l'issue de l'évaluation européenne d'harmonisation dans le cadre d'un accès précoce. Le bénéfice de cette association d'anticorps monoclonaux, en prophylaxie et en phase précoce de la maladie, dans une population à haut risque, est attendu sur la base d'un rationnel pharmacodynamique de thérapie substitutive et des données de plusieurs études de phase 1/2/3.
Ces anticorps monoclonaux font l'objet d'un programme d'accès compassionnel au niveau international. Les données cliniques sont susceptibles d'être ajustées/complétées dans le cadre de la procédure d'évaluation européenne en cours pour cette association. Elles doivent à ce stade, être prises en compte avec prudence.
Traitement de la COVID-19
Données de la Phase 3 de l'étude R10933-10987-COV-2067
Les résultats de l'étude de phase 3 avec un critère de jugement principal clinique ont été rendus disponibles de façon préliminaire et pourront faire l'objet d'un ajustement dans le cadre de la procédure d'évaluation européenne en cours.
L'essai de phase 3, COV-2067, est un essai clinique randomisé, en double aveugle, contrôlé par placebo, évaluant l'association du casirivimab et de l'imdevimab pour le traitement de patients atteints de la COVID-19 qui ne sont pas hospitalisés.
4567 patients présentant au moins un facteur de risque de faire une forme sévère de la COVID-19 ont été randomisés pour recevoir une perfusion intraveineuse (IV) unique de l'association casirivimab et imdevimab aux posologies de 1200 mg (600 mg de chaque anticorps) (n = 838), de 2400 mg (1200 mg de chaque anticorps) (n= 1 529), de 8 000 mg (4 000 mg de chaque anticorps) (n = 700) ou d'un placebo (n = 1 500). Les données issues de la posologie de 8 000 mg ont été analysées de façon descriptive. Les analyses comparatives ont été réalisées pour les patients randomisés de façon concomitante entre les bras de traitements ou les bras placebo.
A l'inclusion, l'âge médian était de 50 ans (avec 13% des sujets âgés de 65 ans ou plus), 52% des patients étaient des femmes, tous les patients présentaient un ou plusieurs facteurs de risque d'évolution vers une forme sévère de la COVID-19. La durée médiane des symptômes avant la randomisation était de 3 jours et la charge virale moyenne à l'inclusion était de 6,69 log10 copies/ml. Les données démographiques et les caractéristiques de la maladie à l'inclusion étaient bien équilibrées entre les différents bras de traitement et les bras placebo.
Le critère d'évaluation principal était la proportion de patients avec au moins une hospitalisation liée à la COVID-19 ou le décès (toutes causes confondues) jusqu'au jour 29. Ce critère d'évaluation principale a été évalué chez des patients qui présentaient lors de la randomisation un test de RT-qPCR pour le SARS-CoV-2 positif (prélèvement nasopharyngé) et avec au moins un facteur de risque d'évolution vers une forme sévère de la COVID-19 (Groupe d'analyse mFAS).
Résultats selon les données préliminaires de l'étude :
Tableau 6: Résumé des principaux résultats de l'étude de Phase III (R10933-10987-COV-2067)
| | 1200 mg IV | Placebo | 2400 mg IV | Placebo |
| n=736 | n=748 | n=1,355 | n=1,341 | |
| Patients avec ≥ 1 hospitalisation liée à la COVID-19 ou décès jusqu'au jour 29 | ||||
| Réduction du risque | 70,4% (p<0,0024) | 71,3% (p<0,0001) | ||
| Nombre de patients avec un évènement | 7 (1,0%) | 24 (3,2%) | 18 (1,3%) | 62 (4,6%) |
| Temps de la résolution des symptômes de la COVID-19 (critère secondaire) | ||||
| Jours médians jusqu'à la résolution des symptômes | 10 | 14 | 10 | 14 |
| Réduction médiane (jours) | 4 (p<0,0001) | 4 (p<0,0001) | ||
Les résultats entre les deux bras de traitements sont également comparables dans les sous- groupes de patients définis par la charge virale ou le statut sérologique à l'inclusion.
Le traitement par l'association casirivimab et imdevimab a entraîné une réduction statistiquement significative de la charge virale moyenne (log10 copies/mL) entre l'inclusion et le jour 7 par rapport au placebo (-0,71 log10 copies/mL pour la posologie de 1200 mg et - 0,86 log10 copies/mL pour la posologie de 2400 mg; p <0,0001). Des réductions ont été observées pour l'ensemble de la population d'analyse mFAS et dans différents sous-groupes de patients, y compris ceux dont la charge virale à l'inclusion était >106 copies/mL ou qui étaient séronégatifs à l'inclusion.
Données de la Phase 3 de l'étude RECOVERY
L'essai RECOVERY (Randomised Evaluation of COVID-19 therapy) est un essai de plateforme ouvert, contrôlé, randomisé destiné à évaluer les effets des traitements potentiels chez les patients hospitalisés pour une infection à SARS-CoV-2. L'essai est en cours dans 181 hôpitaux répartis dans le monde. Parmi ceux-ci, 127 hôpitaux britanniques ont participé à l'évaluation de l'association Casirivimab et Imdevimab.
Etaient éligibles les patients admis à l'hôpital qui présentaient une infection par le SARS- CoV-2 cliniquement suspectée ou confirmée en laboratoire, en l'absence d'antécédents médicaux pouvant faire courir un risque significatif au patient selon l'appréciation du clinicien. Les patients ayant précédemment reçu un traitement par immunoglobuline intraveineuse au cours de l'hospitalisation et les enfants pesant moins de 40 kg ou âgés de moins de 12 ans n'étaient pas admissibles à la randomisation pour l'association Casirivimab et Imdevimab.
La présence d'anticorps anti-SARS-CoV-2 à l'inclusion (IgG anti-Spike) a été déterminée à l'aide d'échantillons de sérum prélevés au moment de la randomisation.
Les patients éligibles ont été randomisés selon un rapport de 1:1 afin de recevoir soit une prise en charge selon les soins standards, soit l'association Casirivimab et Imdevimab associé à une prise en charge selon les soins standards.
Les patients randomisés bénéficiant de l'association Casirivimab et Imdeviamab ont reçu une dose unique de 8000mg de l'association des deux anticorps (casirivimab 4000mg et imdevimab 4000mg) par le biais d'une perfusion intraveineuse sur une durée 60 minutes (+/- 15 minutes). L'administration a été réalisée dès que possible après la randomisation.
Le critère d'évaluation principal était la mortalité à 28 jours, évaluée d'abord chez les patients séronégatifs (IgG anti-Spike) lors de la randomisation, puis dans l'ensemble de la population de l'étude (séronégatifs et séropositifs).
9 785 patients ont été randomisés pour recevoir soit l'association Casirivimab et Imdevimab associé à une prise en charge selon les soins standards soit seulement une prise en charge selon les soins standards. A l'inclusion, 3 153 (32%) patients étaient séronégatifs, 5 272 (54%) étaient séropositifs et 1 360 (14%) avait un statut sérologique inconnu. La majorité des patients était sous oxygénothérapie ou ventilation non-invasive (87%) et ont reçus un traitement par corticoïdes (94%) (voir tableau 7).
Les critères d'évaluation principal et secondaire,chez l'ensemble des patients et en fonction du statut sérologique à l'inclusion, sont présentés dans la figure 8. L'administration de 4000mg de Casirivimab et 4000mg d'Imdevimab en complément des soins standards a réduit la mortalité toutes causes confondues de 20 % chez les patients séronégatifs à l'inclusion, par rapport aux patients recevant uniquement les soins standard. Dans le groupe recevant l'association Casirivimab et Imdevimab, 24 % des patients étaient décédés au jour 28, contre 30 % dans le groupe recevant les soins standards seulement ; risque relatif (RR) : 0,80 avec un intervalle de confiance à 95 % (IC) : 0,70-0,91 ; p=0,001. Ce résultat traduit un bénéfice de 6 décès de moins pour 100 patients séronégatifs ayant reçu l'association Casirivimab et Imdevimab. Le bénéfice clinique survient environ une semaine après le début du traitement par l'association Casirivimab et Imdevimab, ce qui suggère que l'initiation précoce du traitement présente un intérêt bénéfique pour les patients.
Tableau 7: Etude RECOVERY : caractéristiques démographiques selon le statut virologique
| | Patient séronégatifs à l'inclusion | Ensemble des patients | ||
| | Casirivimab + Imdevimab et prise en charge selon les pratiques courantes (n=1633) | Prise en charge selon les pratiques courantes (n=1520) | Casirivimab + Imdevimab et prise en charge selon les pratiques courantes (n=4839) | Prise en charge selon les pratiques courantes (n=4946) |
| Âge (ans) | 63.2 (15.5) | 64.0 (15.2) | 61.9 (14.6) | 61.9 (14.4) |
| <70* | 1054 (65) | 943 (62) | 3389 (70) | 3454 (70) |
| 70 to 79 | 348 (21) | 344 (23) | 936 (19) | 962 (19) |
| ≥80 | 231 (14) | 233 (15) | 514 (11) | 530 (11) |
| Sexe | | | | |
| Homme | 995 (61) | 879 (58) | 3033 (63) | 3095 (63) |
| Femme† | 638 (39) | 641 (42) | 1806 (37) | 1851 (37) |
| Nombre de jours depuis l'apparition des symptômes | 7 (4-10) | 7 (5-9) | 9 (6-12) | 9 (6-12) |
| Sérologie SARS-CoV-2 | | | | |
| Positive | 0 | 0 | 2636 (54) | 2636 (53) |
| Négative | 1633 (100) | 1520 (100) | 1633 (34) | 1520 (31) |
| Manquante | 0 | 0 | 570 (12) | 790 (16) |
| Assistance respiratoire | | | | |
| Pas d'oxygène | 182 (11) | 148 (10) | 332 (7) | 309 (6) |
| Oxygénothérapie simple | 1085 (66) | 995 (65) | 2980 (62) | 3016 (61) |
| Ventilation non-invasive | 332 (20) | 341 (22) | 1244 (26) | 1317 (27) |
| Ventilation mécanique invasive | 34 (2) | 36 (2) | 283 (6) | 304 (6) |
| Traitement par corticoïdes | | | | |
| Oui | 1481 (91) | 1399 (92) | 4530 (94) | 4639 (94) |
| Non | 152 (9) | 118 (8) | 308 (6) | 299 (6) |
| Inconnu | 0 | 3 (<1) | 1 (<1) | 8 (<1) |
Les données sont des moyennes (écart-type), des n (%) ou des médianes (IQR). *Inclut 11 enfants (<18 ans). † Comprend 25 femmes enceintes.
Tableau 8: Etude RECOVERY (résultats préliminaires) : critères d'évaluation principal et secondaire dans l'ensemble de la population de l'étude et chez les patients séronégatifs :
Prévention de la COVID-19 (prophylaxie pré et post-exposition au SARS-CoV-2)
L'essai de phase 3 COV-2069 est un essai clinique randomisé, en double aveugle, contrôlé par placebo, évaluant l'association du casirivimab et de l'imdevimab pour la prévention de la COVID-19 chez les cas-contact familiaux d'individus infectés par le SARS-CoV-2 (cas index).
L'essai a recruté des adultes et adolescents ≥12 ans, non-vaccinés et asymptomatiques pour la COVID-19 qui partagent le même foyer qu'un patient infecté par le SARS-CoV-2. Les individus ont été randomisés 1:1 pour recevoir une dose unique de l'association du casirivimab et de l'imdevimab à la posologie de 1 200 mg (600 mg de casirivimab et 600 mg d'imdevimab) ou un placebo administrés par voie sous-cutanée dans les 96 heures suivant la confirmation d'un test de diagnostic du SARS-CoV-2 positif des cas index. Les individus randomisés ont rejoint la cohorte A ou B de l'étude en fonction du résultat de RT-qPCR pour le SARS-CoV-2à l'inclusion (négatif ou positif).
Étude COV-2069, Cohorte A
2 067 sujets cas-contact avec un résultat de RT-qPCR négatif au SARS-CoV-2 à l'inclusion ont été recrutés et randomisés. La population d'analyse principale comprenait des individus qui étaient SARS-CoV-2 négatifs et séronégatifs à l'inclusion. Les individus qui étaient séropositifs ou qui avaient une sérologie à l'inclusion indéterminée/manquante ont été exclus de l'analyse d'efficacité primaire. Une analyse de sensibilité des résultats indépendamment du statut sérologique à l'inclusion a également été réalisée. Sur les 1 505 participants de la population d'analyse primaire, 753 personnes ont reçus l'association des deux anticorps et 752 personnes ont reçus un placebo. Tous les participants ont par la suite réalisé un test RT- qPCR pour le SARS-CoV-2 via un écouvillon nasopharyngé tous les 7 jours ainsi que des entretiens hebdomadaires avec l'investigateur pour une évaluation des symptômes de la COVID-19 au cours de la période d'évaluation de l'efficacité de 28 jours. Aucune donnée n'a été recueillie sur le type ou l'étendue de l'exposition au cas index.
Pour la population de l'analyse primaire à l'inclusion, l'âge médian était de 44 ans (avec 9 % des participants âgés de 65 ans ou plus), 54 % des individus étaient des femmes. Les caractéristiques démographiques à l'inclusion et les caractéristiques de la maladie étaient bien équilibrées entre les bras de traitement et placebo.
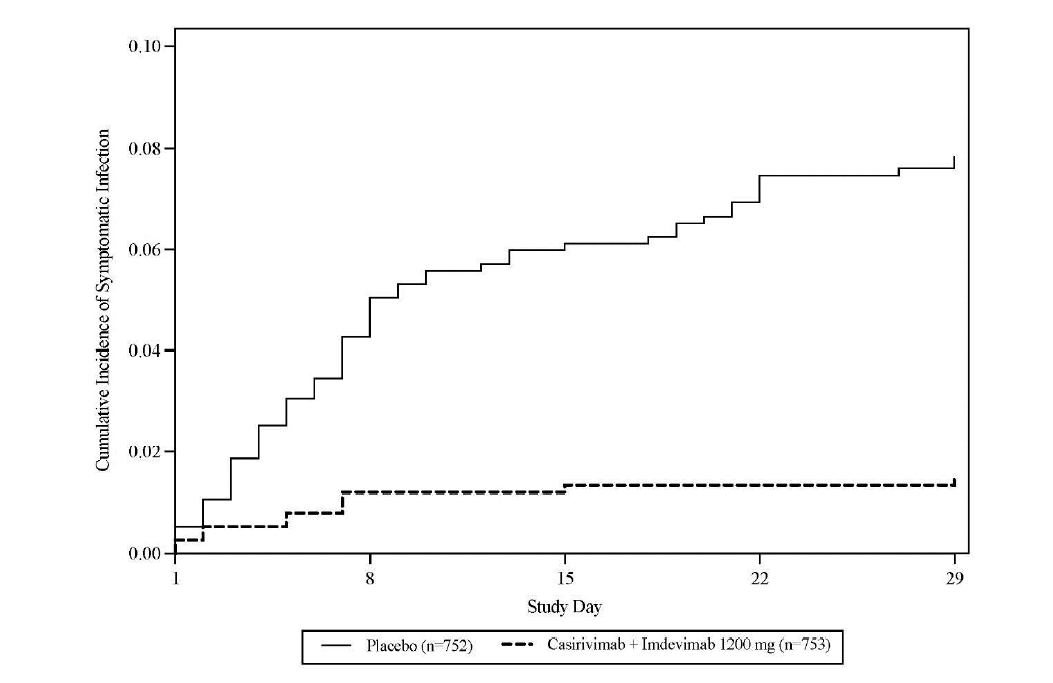
Le critère d'évaluation principal de l'efficacité dans la population d'analyse principale était la proportion de participants qui ont développé une forme symptomatique de la COVID-19 avec une infection au SARS-CoV-2 confirmée par un test RT-qPCR jusqu'au jour 29. Dans cette population, une réduction statistiquement significative de 81 % du risque de développer une forme symptomatique de COVID-19 avec l'association du casirivimab et de l'imdevimab versus placebo a été observée (voir Tableau 9 et Figure 2).
Tableau 9: Principaux résultats de l'essai de phase 3 pour la prévention de la COVID-19 chez des individus cas-contact non infectés par le SARS-CoV-2 à l'inclusion. Étude COV-2069, cohorteA
| | Casirivimab et Imdevimab (dose unique de 1 200 mg, SC) | Placebo |
| Population d'analyse primaire : séronégative à l'inclusion | n = 753 | n = 752 |
| Risque d'une forme symptomatique de la COVID-19 | ||
| Jusqu'au jour 29 (critère d'évaluation principal) | ||
| Réduction de risque (Odds ratio, p-Value) | 81% (0.17; p < 0.0001) | |
| Nombre d'individus avec événements | 11 (1.5%) | 59 (7.8%) |
| Durant la première semaine1 | ||
| Réduction de risque (Odds ratio, nominal p-Value) | 72% (0.27; p = 0.0005) | |
| Number of individuals with events | 9 (1.2%) | 32 (4.3%) |
| Après la première semaine1 | ||
| Réduction de risque (Odds ratio, nominal p-Value) | 93% (0.07; p = 0.0003) | |
| Nombre d'individus avec événements | 2 (0.3%) | 27 (3.6%) |
| Symptômes et charge virale | ||
| Nombre total de semaines avec symptômes (critère secondaire) | ||
| Réduction2 | 93% (p < 0.0001) | |
| Nombre moyen de semaines avec symptômes chez les individus symptomatiques3 | 1.2 | 3.2 |
| Incidence de toute infection au SARS-CoV-2 confirmée par un test RT-qPCR positif (critère secondaire) | ||
| Réduction de risque (Odds ratio, p-Value) | 66% (0.31; p < 0.0001) | |
| Nombre d'individus avec événements | 36 (4.8%) | 107 (14.2%) |
| Nombre total de semaines avec une infection au SARS-CoV-2 (test RT-qPCR positif) quels que soient les symptômes (critère secondaire) | ||
| Réduction2 | 82% (p < 0.0001) | |
| Nombre moyen de semaines avec des individus infectés3 | 1.1 | 2.2 |
| Nombre total de semaines avec une charge virale élevée (>104 copies/mL) (critère secondaire) | ||
| Réduction2 | 90% (p < 0.0001) | |
| Nombre moyen de semaines avec une charge virale élevée chez les individus positifs au SARS-CoV-2 ( RT-qPCR)3 | 0.4 | 1.3 |
| Hospitalisations ou visites aux urgences liées à la COVID-191 | ||
| Réduction de risque (nominal p value) | 100% (p = 0.0621) | |
| Nombre d'individus avec événements | 0 (0%) | 4 (0.53%) |
| | ||
| Tous les participants, quel que soit le statut sérologique à l'inclusion | 1046 | 1021 |
| Risque de la COVID-19 jusqu'au jour 29 (analyse de sensibilité)3 | ||
| Réduction de risque (Odds ratio, nominal p-value) | 82% (0.17; p < 0.0001) | |
| Nombre d'individus avec événements | 12 (1.1%) | 66 (6.5%) |
| | ||
| Séropositif à l'inclusion | 235 | 222 |
| Risque de la COVID-19 jusqu'au jour 293 | ||
| Réduction de risque (Odds ratio, nominal p-value) | 81% (0.19; p = 0.1369) | |
| Nombre d'individus avec événements | 1 (0.4%) | 5 (2.3%) |
1. Ces analyses ne faisaient pas partie du plan d'analyse statistique pré-planifié, les valeurs p sont donc nominales
2. Basé sur la durée normalisée pour 1000 sujets
3. Pré-spécifié mais non inclus dans la hiérarchie des tests d'hypothèse
4. Seuls les participants ayant une charge virale post-inclusion ont été inclus
Figure 2: COV-2069-A - Incidence cumulative d'une forme symptomatique de la COVID-19 par jour d'étude
Dans une analyse post-hoc pour les sous-groupes de participants qui répondaient aux critères de risque élevé de progression vers une forme sévère de la COVID-19, il y avait une réduction de 76% du risque de développer une forme symptomatique de la COVID-19 avec le traitement par casirivimab et imdevimab versus placebo [10/570 (2%) vs 42/567 (7 %) ; odds ratio ajustés 0,22 ; p < 0,0001].
Étude COV-2069, Cohorte B
314 sujets cas-contact avec un résultat de RT-qPCR positif au SARS-CoV-2 à l'inclusion ont été recrutés et randomisés. La population d'analyse principale comprenait des individus asymptomatiques qui étaient infectés par le SARS-CoV-2 (RT-qPCR positif) et séronégatifs à l'inclusion. Sur les 204 participants de la population d'analyse primaire, 100 personnes ont reçus l'association casirivimab et imdevimab et 104 personnes ont reçus un placebo. Tous les participants ont par la suite réalisé un test de RT-qPCR pour le SARS-CoV-2 via un écouvillon nasopharyngé tous les 7 jours ainsi que des entretiens hebdomadaires avec l'investigateur pour une évaluation des symptômes de la COVID-19 au cours de la période d'évaluation de l'efficacité de 28 jours. Aucune donnée n'a été recueillie sur le type ou l'étendue de l'exposition au cas index.
Pour la population de l'analyse primaire à l'inclusion, l'âge médian était de 40 ans (avec 11 % des participants âgés de 65 ans ou plus), 55 % des individus étaient des femmes. Les caractéristiques démographiques à l'inclusion et les caractéristiques de la maladie étaient bien équilibrées entre les bras de traitement par casirivimab et imdevimab et placebo.
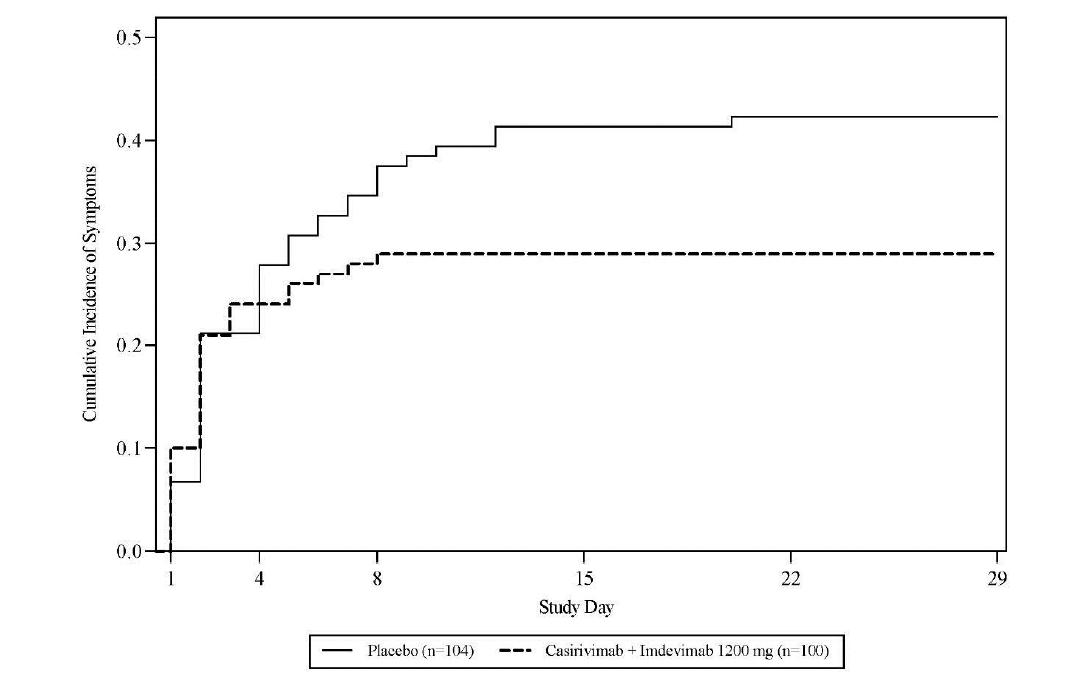
Le critère d'évaluation principal d'efficacité dans la population d'analyse principale était la proportion de patients qui ont développé une forme symptomatique de la COVID-19 (confirmée par un test RT-qPCR) jusqu'au jour 29. Une réduction de 31 % du risque de développer des symptômes de la COVID-19 a été observée (voir Tableau 10 et Figure 3).
Tableau 10: Principaux résultats de l'étude COV-2069 chez les patients infectés par le SARS- CoV-2 à l'inclusion et asymptomatiques (cohorte B)
| | Casirivimab et Imdevimab (Dose unique de 1 200 mg) | Placebo |
| Population d'analyse primaire : séronégative à l'inclusion | n = 100 | n = 104 |
| Risque d'une forme symptomatique de la COVID-19 | ||
| Réduction globale du risque jusqu'au jour 29 (critère d'évaluation principal) | ||
| Réduction de risque (Odds ratio, p-Value) | 31% (0.54; p = 0.0380) | |
| Nombre d'individus avec événements | 29 (29%) | 44 (42.3%) |
| Réduction globale du risque après le jour 3 (jours 4-29)1,2 | ||
| Réduction de risque (Odds ratio, nominal p-Value) | 76% (0.18; p = 0.0010) | |
| Nombre d'individus avec événements | 5 (5%) | 22 (21.2%) |
| Symptômes et charge virale | ||
| Nombre total de semaines avec symptômes (critère d'évaluation secondaire) | ||
| Réduction (p-Value) | 45% (p = 0.0273) | |
| Nombre total de semaines (cumulé pour tous les patients dans chaque bras) | 90 | 170 |
| Nombre total de semaines avec une charge virale élevée (>104 copies/mL) (critère secondaire) | ||
| Réduction (p-Value) | 40% (p = 0.0010) | |
| Nombre total de semaines (cumulé pour tous les patients dans chaque bras) | 48 | 82 |
| Hospitalisations ou visites aux urgences liées à la COVID-192 | ||
| Réduction de risque (nominal p-value) | 100% (p = 0.0292) | |
| Nombre d'individus avec événements | 0 (0%) | 6 (5.8%) |
| | ||
| Tous les sujets quel que soit le statut sérologique | n = 155 | n = 156 |
| Risque d'une forme symptomatique de la COVID-19 jusqu'au jour 29 (analyse de sensibilité) | ||
| Réduction globale du risque jusqu'au jour 29 | ||
| Réduction de risque (Odds ratio, nominal p-value) | 35% (0.54; p = 0.0166) | |
| Nombre d'individus avec événements | 34 (21.9%) | 53 (34%) |
1. N'inclut pas les résultats des jours 1 à 3, lorsque les événements étaient similaires entre les groupes de traitement
2. Ces analyses ne faisaient pas partie du plan d'analyse statistique pré-planifié, les valeurs p sont donc nominales
Figure 3: Incidence cumulative d'une forme symptomatique de la COVID-19 par jour d'étude COV-2069, Cohort B
Dans une analyse post-hoc réalisée dans les différents sous-groupe de patients qui répondaient aux critères de risque élevé de progression vers une forme sévère de la COVID- 19, une réduction non-statistiquement significative de 28% du risque de développer une forme symptomatique de la COVID-19 avec le traitement par casirivimab et imdevimab a été observée par rapport au placebo [22/72 (31%) vs 31/73 (43 %) ; odds ratio ajusté de 0,60 ; p = 0,1877].
Étude HV-2093
Les données soutenant l'utilisation en dose répétées de l'association du casirivimab et de l'imdevimab en prophylaxie pré-exposition pour la prévention de la COVID-19 sont basées sur l'analyse exploratoire des données d'efficacité et de pharmacocinétique issues de 969 participants de la phase 1 HV-2093. L'étude HV-2093 est un essai clinique de phase 1 randomisé, en double aveugle, contrôlé par placebo, évaluant l'innocuité, la tolérance, la pharmacocinétique et l'immunogénicité de doses sous-cutanées répétées (jusqu'à 6 doses sur 24 semaines) de l'association casirivimab et imdevimab chez des participants adultes, négatifs pour le SARS-CoV-2 à l'inclusion. Les individus ont été randomisés 3:1 pour recevoir des injections sous-cutanées toutes les 4 semaines pendant 24 semaines de 1 200 mg de l'association casirivimab et imdevimab (600 mg de casirivimab et 600 mg d'imdevimab) (n = 729) ou un placebo (n = 240).
A l'inclusion, l'âge médian était de 47 ans (avec 13 % des participants âgés de 65 ans ou plus), 55 % des individus étaient des hommes. Les données démographiques à l'inclusion et les caractéristiques de la maladie étaient bien équilibrées entre les groupes de traitement et placebo.
L'objectif principal de l'étude était la pharmacocinétique (voir rubrique Propriétés pharmacocinétiques). Un critère d'évaluation de l'efficacité était l'incidence de la COVID-19 cliniquement diagnostiquée. Au cours de la période de traitement de six mois, une réduction de 92% du risque de développer des symptômes de la COVID-19 avec le traitement par casirivimab et imdevimab a été observée par rapport au placebo : 3/729 (0,4%) pour le bras de traitement contre 12/240 (5,0%) pour le placebo ; odds ratio (OR) 0,08 (IC à 95 % : 0,01, 0,30) ; p nominal < 0,0001. Parmi les participants qui ont développé une forme symptomatique de la COVID-19, 9/12 individus ayant reçu le placebo avaient un résultat de RT-PCR positif au SARS-CoV-2 ou étaient séropositifs alors que 0/3 participants du groupe casirivimab et imdevimab avaient un résultat de RT-PCR positif au SARS-CoV-2 ou étaient séropositifs à la fin de la période de traitement.
Immunogénicité
Comme pour toutes les protéines thérapeutiques, il existe un risque potentiel d'immunogénicité. La détection de la formation d'anticorps dépend fortement de la sensibilité et de la spécificité des tests réalisés. De plus, l'incidence observée de la positivité des anticorps (y compris les anticorps neutralisants) dans un test peut être influencée par plusieurs facteurs, notamment la méthodologie du test, la manipulation des échantillons, le moment du prélèvement des échantillons, les médicaments concomitants et la maladie sous- jacente. Pour ces raisons, la comparaison de l'incidence des anticorps anti-casirivimab et anti-imdevimab dans les études décrites ci-dessous avec l'incidence décrite dans d'autres études ou avec d'autres thérapeutiques peut être source de discordances.
Chez tous les individus ayant reçu l'association casirivimab et imdevimab soit par perfusion intraveineuse ou par injection sous-cutanée, l'incidence des anticorps anti-casirivimab et anti-imdevimab était respectivement de 0,8 % et 1,7 %. Pour les individus ayant reçu un placebo, l'incidence des anticorps anti-casirivimab et anti-imdevimab était respectivement de 1,9 % et 4,5 %.
Chez 707 individus traités par l'association casirivimab et imdevimab à la posologie de 1200 mg (600 mg de casirivimab et 600 mg d'imdevimab) par voie sous-cutanée toutes les 4 semaines, l'incidence des anticorps anti-casirivimab et anti-imdevimab apparus pendant le traitement était respectivement de 0,1 % et 2,0 %. Parmi les 232 individus ayant reçus des doses répétées de placebo, l'incidence des anticorps anti-casirivimab et anti-imdevimab apparus sous traitement était respectivement de 0 % et 2,6 %. Les titres d'anticorps chez les individus recevant des doses répétées de l'association du casirivimab et de l'imdevimab et du placebo étaient faibles, sans aucun signe de modification des profils pharmacocinétiques du casirivimab ou de l'imdevimab.
Absorption
Après une administration en dose unique par voie IV, le casirivimab et l'imdevimab ont tous deux présenté une pharmacocinétique linéaire et proportionnelle pour des doses allant de 300 mg (150 mg de casirivimab et 150 mg d'imdevimab) à 8000 mg (4000 mg de casirivimab et 4000 mg d'imdevimab).
Le tableau 11 présente un résumé des paramètres pharmacocinétiques du casirivimab et de l'imdevimab, après l'administration d'une dose unique par voie IV de 600 mg de casirivimab et 600 mg d'imdevimab, calculés à l'aide d'un modèle pharmacocinétique de population pour chaque anticorps basé sur les données de 3687 individus pour le casirivimab et 3716 individus pour l'imdevimab.
Tableau 11: Résumé des paramètres pharmacocinétiques du casirivimab et de l'imdevimab après l'administration d'une dose unique par voie IV de 1200 mg (600 mg de casirivimab et 600 mg d'imdevimab)
| Paramètre pharmacocinétique1 | casirivimab | imdevimab |
| AUC0-28 (mg·jour/L)2 | 1754.9 (380.50) | 1600.8 (320.88) |
| AUCinf (mg·jour/L)3 | 3563.6 (1239.61) | 2890.5 (876.31) |
| Cmax (mg/L)4 | 182.7 (81.45) | 181.7 (77.78) |
| C28 (mg/L)5 | 37.9 (10.33) | 31.0 (8.24) |
| Demi-vie (jour) | 31.2 (10.59) | 27.3 (7.73) |
1 Moyenne et écart type de la moyenne; 2 AUC0-28 = Aire sous la courbe concentration-temps de t0 au jour 28 ; 3 AUCinf = Aire sous la courbe concentration-temps de t0 à l'infini; 4 Cmax = Concentration maximale dans le sérum et représente la concentration à la fin de la perfusion; 5 C28 = Concentration au jour 28 après administration ; Cmax = Concentration sérique maximale
Le tableau 12 présente un résumé des paramètres pharmacocinétiques du casirivimab et de l'imdevimab, après l'administration d'une dose unique par voie SC de 600 mg de casirivimab et 600 mg d'imdevimab, calculés à l'aide d'un modèle pharmacocinétique de population pour chaque anticorps.
Tableau 12: Résumé des paramètres pharmacocinétiques du casirivimab et de l'imdevimab après l'administration d'une dose unique par voie SC de 1200 mg (600 mg de casirivimab et 600 mg d'imdevimab)
| Paramètre pharmacocinétique1 | casirivimab | imdevimab |
| AUC0-28 (mg·jour/L)2 | 1121.7 (243.12) | 1016.9 (203.92) |
| AUCinf (mg·jour/L)3 | 2559.5 (890.35) | 2073.3 (628.60) |
| Cmax (mg/L)4 | 52.2 (12.15) | 49.2 (11.01) |
| tmax (jour)5, 6 | 6.7 [3.4, 13.6] | 6.6 [3.4, 13.6] |
| C28 (mg/L)7 | 30.5 (7.55) | 25.9 (6.07) |
1 Moyenne et écart type de la moyenne; 2 AUC0-28 = Aire sous la courbe concentration-temps de t0 au jour 28 ; 3 AUCinf = Aire sous la courbe concentration-temps de t0 à l'infini ;4 Cmax = Concentration sérique maximale; 5 tmax= temps pour atteindre la Cmax; 6 Médiane [minimum, maximum]; 7 C28 = Concentration au jour 28 après administration i.e., au jour 29;
Le tableau 13 présente un résumé des paramètres pharmacocinétiques du casirivimab et de l'imdevimab, après l'administration d'une dose initiale, par voie IV, de 600 mg de casirivimab et 600 mg d'imdevimab, suivie par des administrations IV, répétées toutes les 4 semaines, de 300 mg de casirivimab et 300 mg d'imdevimab, calculés à l'aide d'un modèle pharmacocinétique de population pour chaque anticorps.
Tableau 13: Résumé des paramètres pharmacocinétiques de casirivimab et d'imdevimab après administration d'une dose initiale par voie IV de 1200 mg (600 mg de casirivimab et 600 mg d'imdevimab) suivi par des doses IV de maintenance de 600 mg (300 mg de casirivimab et 300 mg d'imdevimab)
| Paramètre pharmacocinétique1 | casirivimab | imdevimab |
| AUCtau,ss (mg∙jour/L)2 | 1767.5 (605.79) | 1436.8 (432.87) |
| Cmax,ss (mg/L)3 | 133.8 (46.51) | 122.4 (41.67) |
| Ctrough,ss (mg/L)4 | 42.6 (19.72) | 31.7 (13.56) |
| C28 (mg/L)5 | 37.9 (10.32) | 31.0 (8.24) |
| AR6 | 1.0 (0.241) | 0.893 (0.174) |
1 Moyenne et écart type de la moyenne; 2 AUCtau,ss = Aire sous la courbe concentration-temps durant un intervalle de dose à l'état d'équilibre; 3Cmax ss = Concentration sérique maximale à l'état d'équilibre; 4Ctrought ss = Concentration sérique minimale à l'état d'équilibre; 5 C28 = Concentration au jour 28 après administration de la première dose; 6 AR = ratio d'accumulationa
Note: a le ratio d'accumulation est calculé de la manière suivant : (FD = première dose).
Le tableau 14 présente un résumé des paramètres pharmacocinétiques du casirivimab et de l'imdevimab, après l'administration d'une dose initiale, par voie SC, de 600 mg de casirivimab et 600 mg d'imdevimab, suivie par des administrations SC, répétées toutes les 4 semaines, de 300 mg de casirivimab et 300 mg d'imdevimab, calculés à l'aide d'un modèle pharmacocinétique de population pour chaque anticorps.
Tableau 14: Résumé des paramètres pharmacocinétiques du casirivimab et de l'imdevimab après l'administration d'une dose initiale par voie SC de 1200 mg (600 mg de casirivimab et 600 mg d'imdevimab) suivi par des doses SC de maintenance de 600 mg (300 mg de casirivimab et 300 mg d'imdevimab)
| Paramètre pharmacocinétique1 | casirivimab | imdevimab |
| AUCtau,ss (mg∙jour/L)2 | 1268.9 (434.68) | 1030.1 (310.30) |
| Cmax,ss (mg/L)3 | 56.0 (16.81) | 47.0 (12.43) |
| Ctrough,ss (mg/L)4 | 34.0 (14.56) | 26.1 (10.17) |
| C28 (mg/L)5 | 30.5 (7.55) | 25.9 (6.07) |
| AR6 | 1.13 (0.288) | 1.01 (0.213) |
1 Moyenne et écart type de la moyenne; 2 AUCtau,ss = Aire sous la courbe concentration-temps durant un intervalle de dose à l'état d'équilibre;3Cmax ss = Concentration sérique maximale à l'état d'équilibre; 4Ctrought ss = Concentration sérique minimale à l'état d'équilibre; 5 C28 = Concentration au jour 28 après administration de la première dose; 6 AR = ratio d'accumulationa
Note: a le ratio d'accumulation est calculé de la manière suivant : (FD = première dose).
Pour l'administration IV et SC de doses répétées en prophylaxie, les simulations de pharmacocinétiques de population prévoient que les médianes des concentrations sériques minimales à l'état d'équilibre prédites du casirivimab et de l'imdevimab soient similaires aux concentrations sériques moyennes observées au jour 29 pour une dose unique de 1200 mg de l'association des deux anticorps par voie sous-cutanée.
Distribution
Le volume total de distribution estimé par une analyse pharmacocinétique de population est de 7,161 L pour le casirivimab et de 7,425 L pour l'imdevimab.
Biotransformation
Les voies métaboliques du casirivimab et de l'imdevimab n'ont pas été caractérisées. En tant qu'anticorps monoclonaux humains d'isotype IgG1, le casirivimab et l'imdevimab devraient être dégradés en petits peptides et acides aminés via les voies cataboliques de la même manière que les IgG endogènes.
Elimination
D'après une analyse pharmacocinétique de population, la demi-vie et la clairance du casirivimab et de l'imdevimab sont présentées dans le tableau 15.
Tableau 15: Demi-vie et clairance plasmatique du casirivimab et de l'imdevimab après l'administration de doses IV uniques
| | Casirivimab | Imdevimab | ||
| Paramètre pharmacocinétique | Moyenne | Intervalle de confiance à 95% | Moyenne | Intervalle de confiance à 95% |
| Demi-vie (jour) | 29,8 | (16,4 ; 43,1) | 26,2 | (16,9 ; 35,6) |
| Clairance plasmatique (L/jour) | 0,188 | (0,11 ; 0,3) | 0,227 | (0,15 ; 0,35) |
Le casirivimab et l'imdevimab sont des anticorps monoclonaux et ne sont donc pas susceptibles d'être excrétés par voie rénale.
Population pédiatrique
Des adolescents (≥12 ans et ≥40 kg) ont été inclus dans les études (COV-2067, COV-2069) cependant aucune donnée pharmacocinétique n'était disponible chez ces individus. Étant donné que l'intervalle de poids corporel des adolescents se situe généralement dans l'intervalle de poids des sujets adultes et que généralement le poids est la principale covariable affectant l'exposition au produit dans cette tranche d'âge, les expositions du casirivimab et de l'imdevimab chez les individus adolescents (≥40kg) devraient être similaires à celles des adultes. La pharmacocinétique du casirivimab et de l'imdevimab chez les patients pédiatriques (<12 ans) n'a pas été établie.
Populations spécifiques
Les effets de l'âge, de l'insuffisance rénale ou de l'insuffisance hépatique sur la pharmacocinétique du casirivimab et de l'imdevimab sont inconnus. L'insuffisance rénale ne devrait pas avoir d'impact sur la pharmacocinétique du casirivimab et de l'imdevimab, étant donné que les anticorps monoclonaux de poids moléculaire >50 kDa ne sont pas connus pour être éliminés par voie rénale. De même, la dialyse ne devrait pas avoir d'incidence sur la pharmacocinétique du casirivimab et de l'imdevimab.
Patients âgés
Il existe peu de données sur l'efficacité et la tolérance du casirivimab et de l'imdevimab chez les patients âgés de 65 ans et plus. Sur les 4567 patients ambulatoires atteints d'une infection par le SARS-CoV-2 qui ont été randomisés dans l'essai clinique R10933-10987- COV-2067, 14% des patients avaient 65 ans ou plus et 4% des patients étaient âgés de 75 ans ou plus. La pharmacocinétique du casirivimab et de l'imdevimab chez les patients âgés par rapport aux patients plus jeunes est inconnue.
L'association de casirivimab et l'imdevimab n'a aucun effet ou un effet négligeable sur l'aptitude à conduire des véhicules et à utiliser des machines. Cependant, certains des effets indésirables mentionnés dans la rubrique Effets indésirables peuvent altérer temporairement l'aptitude à conduire des véhicules ou à utiliser des machines.
Les profils de toxicologie non clinique de l'association casirivimab et imdevimab ont été caractérisés par la conduite d'une étude de toxicologie à doses répétées chez des singes cynomolgus. Ces deux anticorps monoclonaux ont été administrés chaque semaine, seuls (50 mg / kg) par injection en bolus IV, et en combinaison (jusqu'à 150 mg / kg / anticorps) par injection IV ou SC. L'administration une fois par semaine de casirivimab, d'imdevimab, ou de l'association casirivimab + imdevimab a été bien tolérée à toutes les doses, sans effet toxique évident (lié ou non au traitement) pendant la période d'administration de 4 semaines ou au moment de l'autopsie des animaux. Des augmentations transitoires mineures de l'AST et de l'ALT ont été observées avec le casirivimab et l'imdevimab.
Une étude de réactivité croisée des tissus ex vivo a été menée à l'aide d'échantillons de tissus humains sains et de singes cynomolgus. Il n'y avait pas de marquage non spécifique du casirivimab ou de l'imdevimab dans aucun des tissus humains ou de singe utilisés, ce qui était attendu car ces deux anticorps sont dirigés contre une protéine exogène.
Pour les indications de traitement à dose unique, lorsque l'AUCcum estimée pour la dose maximale sans effet néfaste observable dans l'étude de toxicologie de 4 semaines est comparée à l'AUCinf prédite chez les sujets humains, les multiples d'exposition sont d'environ 37,5 pour l'association casirivimab et imdevimab à la posologie de 1 200 mg (600 mg mg de casirivimab et 600 mg d'imdevimab) par voie IV et et 52,3 par voie sous-cutanée.
Pour les indications de prévention chronique à doses répétées, lorsque l'AUC estimée à 4 semaines après la dernière dose pour la dose maximale sans effet néfaste observable dans l'étude de toxicologie de 4 semaines (~AUC tau, ss) est comparée à l'AUCtau, ss prédite chez les sujets humains, les multiples d'exposition sont d'environ 35,3 pour l'association casirivimab et imdevimab à la posologie de 1 200 mg (600 mg de casirivimab et 600 mg d'imdevimab) par voie IV et 49,1 pour la posologie 1 200 mg (600 mg de casirivimab et 600 mg d'imdevimab) de la dose initiale sous-cutanée, suivis des doses de maintenance de 600 mg (300 mg de casirivimab et 300 mg d'imdevimab) toutes les 4 semaines.
Génotoxicité/Cancérogénicité :
Aucune étude de génotoxicité ou de cancérogénicité n'a été effectuée chez l'animal pour évaluer le potentiel génotoxique et cancérogène du casirivimab et de l'imdevimab.
Toxicité de la reproduction et du développement:
Il n'y a pas eu d'étude réalisée chez l'animal permettant d'évaluer les effets du casirivimab et de l'imdevimab sur la toxicologie de la reproduction et sur la fertilité
L'association casirivimab et imdevimab doit être administrée par voie IV ou SC uniquement.
Préparation à l'administration d'une perfusion intraveineuse
Instructions pour la dilution
L'association casirivimab et imdevimab doit être préparée par un professionnel de santé en utilisant une technique aseptique :
1. Sortir les flacons de casirivimab et d'imdevimab du réfrigérateur et les laisser atteindre la température ambiante pendant environ 20 minutes avant leur préparation. Ne pas exposer à une source de chaleur directe. Ne pas agiter les flacons.
2. Inspectez visuellement les flacons de casirivimab et d'imdevimab à la recherche de particules ou d'un changement de couleur avant de les administrer. Si l'un ou l'autre est observé, la solution à diluer doit être jetée et un nouveau flacon doit être utilisé. La solution à diluer de chaque flacon doit être transparente à légèrement opalescente, incolore à jaune pâle.
3. Procurez-vous une poche pour perfusion IV pré-remplie contenant soit 50 mL, 100 mL,
150 mL ou 250 mL de chlorure de sodium injectable à 0,9 %. Dans le cas de l'administration d'une dose de 4000mg de casirivimab et 4000mg d'imdevimab, la dilution devra être uniquement effectuée dans une poche de pour perfusion IV pré-remplie contenant 250 mL de chlorure de sodium injectable à 0,9 % (voir tableau 18).
4. Prélevez le volume nécessaire de casirivimab et d'imdevimab dans chaque flacon respectif à l'aide de deux seringues séparées pour chaque prélèvement et injecter l'intégralité du produit dans la poche pour perfusion pré-remplie contenant la solution de chlorure de sodium injectable à 0,9%, (voir tableau 16). Jetez tout produit restant dans les flacons.
5. Retournez manuellement la poche pour perfusion environ 10 fois pour la mélanger. Ne pas agiter. Ce produit ne contient aucun conservateur, en conséquence, la solution diluée pour perfusion doit être administrée immédiatement après sa préparation.
En cas d'utilisation non immédiate, les durées et conditions de conservation en cours d'utilisation relèvent de la responsabilité de l'utilisateur et ne devraient normalement pas dépasser 24 heures à 2-8°C, sauf si la méthode de dilution prévient tout risque de contamination microbienne. Si elle est réfrigérée, laisser la solution de perfusion revenir à la température ambiante pendant environ 30 minutes avant de l'administrer.Tableau 16: Recommandations pour la dilution de casirivimab et imdevimab pour perfusion IV
| Quantité totale pour 1 dose | Volume total pour 1 dose | Volume de produit prélevé à injecter dans une poche pour perfusion préremplie de chlorure de sodium à 0,9 % | |
|
|
|
|
2.5 mL de
deux flacons unidoses de
casirivimab 2,5 mL de deux flacons unidoses d'imdevimab |
| Dose unique : Traitement Patient ne nécessitant pas d'oxygénothérapie du fait de la COVID-19 | 600 mg casirivimab et 600 mg imdevimab | 10 mL | |
| 5,0 mL d'un flacon multidose de casirivimab 5.0 mL d'un flacon multidose d'imdevimab | |||
|
2,5 mL
de deux flacons unidoses de
casirivimab 5,0 mL d'un flacon multidose d'imdevimab | |||
| 5,0 mL d'un flacon multidose de casirivimab 2,5 mL de deux flacons unidoses d'imdevimab | |||
|
|
|
| |
| Dose unique : Traitement Patient hospitalisé du fait de la COVID-19 et séronégatif nécessitant une oxygénothérapie non invasive. | 4000 mg casirivimab et 4000 mg imdevimab | 66,6 mL |
11,1 mL
de trois flacons multidoses de
casirivimab 11,1 mL de trois flacons multidoses d'imdevimab |
| Dose unique : Prophylaxie post- exposition ou Dose initiale : Prophylaxie pré- exposition | 600 mg casirivimab et 600 mg imdevimab | 10 mL |
2.5 mL de
deux flacons unidoses de
casirivimab 2,5 mL de deux flacons unidoses d'imdevimab |
| 5,0 mL d'un flacon multidose de casirivimab 5.0 mL d'un flacon multidose d'imdevimab | |||
|
2,5 mL de
deux flacons unidoses de
casirivimab 5,0 mL d'un flacon multidose d'imdevimab | |||
|
5,0 mL d'un flacon multidose de casirivimab 2,5 mL de deux flacons unidoses d'imdevimab | |||
|
Dose
répétée de maintenance en prophylaxie pré- exposition a |
300 mg casirivimab et 300 mg imdevimab | 5 mL | 2,5 mL d'un flacon unidose de
casirivimab 2,5 mL d'un flacon unidose d'imdevimab |
| 2,5 mL d'un flacon multidose de casirivimab 2,5 mL d'un flacon multidose de 20 mL d'imdevimab | |||
|
|
|
2,5 ml d'un flacon unidose de casirivimab 2,5 mL d'un flacon multidose d'imdevimab | |
|
|
|
2,5 mL d'un flacon multidose de casirivimab 2,5 mL d'un flacon unidose d'imdevimab |
a Dose subséquente à l'administration de la dose initiale
Tableau 17 : Modalités d'administration recommandées pour l'association de casirivimab et d'imdevimab en perfusion intraveineuse lors de l'utilisation des posologies de 300mg ou 600mg de casirivimab et 300mg ou 600mg d'imdevimab.
| Taille de la poche de perfusion préremplie de chlorure de sodium à 0,9 % | Débit de perfusion maximal | Durée minimale de la perfusion |
| 50 mL | 210 mL/hr | 20 minutes |
| 100 mL | 360 mL/hr | 20 minutes |
| 150 mL | 510 mL/hr | 20 minutes |
| 250 mL | 540 mL/hr | 30 minutes |
Tableau 18 : Modalité d'administration recommandée pour l'association de casirivimab et d'imdevimab en perfusion intraveineuse lors de l'utilisation de la posologie de 4000mg de casirivimab et 4000mg d'imdevimab.
| Taille de la poche de perfusionpréremplie de chlorure de sodium à 0,9 % | Débit de perfusion maximal | Durée minimale de la perfusion |
| 250 mL | 250 mL/hr | 60 minutes (+/-15mn) |
Le casirivimab et l'imdevimab sont à administrer ensemble en une seule perfusion intraveineuse.
Une fois la perfusion terminée, rincer avec au minimum 25 à 50 mL de solution de chlorure de sodium à 9 mg/mL (0,9 %).
Instruction pour l'administration IV
· La solution pour perfusion contenant l'association casirivimab et imdevimab doit être administrée par un professionnel de santé qualifié en utilisant une technique aseptique.
· Rassemblez le matériel nécessaire à l'infusion :
o Set de perfusion en chlorure de polyvinyle (PVC), PVC avec revêtement en polyéthylène (PE) ou polyuréthane (PU).
o Filtre en ligne ou d'apoint de 0,2 micron en polyéthersulfone (PES)
· Fixez le dispositif de perfusion à la poche à perfusion.
· Amorcez le dispositif de perfusion.
· Administrez la totalité de la solution pour perfusion contenue dans la poche au moyen d'une pompe ou par gravité via une ligne intraveineuse contenant un filtre stérile en ligne ou d'appoint de 0,2 micron en polyéthersulfone (PES) (voir tableau 17 et 18). En raison du risque de sur-remplissage des poches de sérum physiologique pré remplies, la totalité de la solution de perfusion contenue dans la poche doit être administrée pour éviter un sous-dosage.
· La solution de perfusion préparée ne doit pas être administrée conjointement avec un autre médicament. La compatibilité du casirivimab et de l'imdevimab injectable avec des solutions IV et des médicaments autres que le chlorure de sodium à 0,9 % pour injection n'est pas connue.
· Une fois la perfusion terminée, rincer la tubulure avec du chlorure de sodium injectable à 0,9 % pour assurer l'administration de la dose requise.
· Jeter le produit non utilisé.
· Assurez une surveillance clinique des patients pendant l'administration et observez-les pendant au moins une heure après la fin de la perfusion.
Préparation à l'administration par voie SC
Instructions pour la préparation des seringues
1. Sortez les flacons de casirivimab et d'imdevimab du réfrigérateur et laissez-les atteindre la température ambiante pendant environ 20 minutes avant leur préparation. Ne pas exposer à une source de chaleur directe. Ne pas agiter les flacons.
2. Inspectez visuellement les flacons de casirivimab et d'imdevimab à la recherche de particules ou d'un changement de couleur avant de les administrer. Si l'un ou l'autre est observé, la solution à diluer doit être jetée et un nouveau flacon doit être utilisé. La solution à diluer de chaque flacon doit être transparente à légèrement opalescente, incolore à jaune pâle.
3. L'administration de casirivimab et imdevimab doit être préparée en utilisant le nombre approprié de seringues (voir Tableau 19). Il convient de se procurer des seringues Luer Lock en polypropylène de 3 ou 5 mL avec connexion Luer et des aiguilles de transfert de 21 gauge.
4. Prélevez 2,5 mL dans chaque seringue (voir Tableau 19) pour un total de 4 seringues pour la dose totale combinée de 1200 mg et un total de 2 seringues pour la dose totale combinée de 600 mg. Conserver le produit restant comme indiqué.
5. Remplacez l'aiguille de transfert de 21 gauge par une aiguille de 25 ou 27 gauge pour réaliser l'injection SC.
6. Ce produit est sans conservateur, par conséquent, les seringues préparées doivent être administrées immédiatement.
En cas d'utilisation non immédiate, les durées et conditions de conservation en cours d'utilisation relèvent de la responsabilité de l'utilisateur et ne devraient normalement pas dépasser 24 heures à 2-8°C, sauf si la méthode de dilution prévient tout risque de contamination microbienne. Si elles sont réfrigérées, laisser les seringues s'équilibrer à la température ambiante pendant environ 10 à 15 minutes avant l'administration.
Tableau 19: Recommandations pour la préparation de casirivimab et imdevimab pour administration SC
| Quantité totale pour 1 dose | Volume total pour 1 dose | Volume de produit prélevé pour la préparation de 4 seringues | |
| Dose unique : Prophylaxie post- exposition ou Dose initiale : Prophylaxie pré- exposition | 600 mg casirivimab et 600 mg imdevimab | 10 mL | 2.5 mL de deux flacons unidoses de casirivimab 2,5 mL de deux flacons unidoses d'imdevimab |
| 2,5 mL (2x) d'un flacon multidose de casirivimab 2.5 mL (2x) d'un flacon multidose d'imdevimab | |||
| 2,5 mL de deux flacons unidoses de casirivimab 2.5 mL (2x) d'un flacon multidose d'imdevimab | |||
| 2,5 mL (2x) d'un flacon multidose de casirivimab 2,5 mL de deux flacons unidoses d'imdevimab |
| Quantité totale pour 1 dose | Volume total pour 1 dose | Volume de produit prélevé pour la préparation de 2 seringues | |
| Dose répétée de maintenance en prophylaxie pré- exposition a | 300 mg casirivimab et 300 mg imdevimab | 5 mL | 2,5 mL d'un flacon unidose de casirivimab 2,5 mL d'un flacon unidose d'imdevimab |
| 2,5 mL d'un flacon multidose de casirivimab 2,5 mL d'un flacon multidose d'imdevimab | |||
| 2,5 ml d'un flacon unidose de casirivimab 2,5 mL d'un flacon multidose d'imdevimab | |||
| 2,5 mL d'un flacon multidose de casirivimab 2,5 mL d'un flacon unidose d'imdevimab |
a Dose subséquente à l'administration de la dose initiale
Instruction pour l'administration SC
1. Pour l'administration d'une dose de 1200 mg (600 mg de casirivimab et 600 mg d'imdevimab), rassemblez les 4 seringues (voir Tableau 19) nécessaires à l'administration SC.
2. Pour l'administration de la dose de 600 mg (300 mg de casirivimab et 300 mg d'imdevimab), rassemblez les 2 seringues (voir Tableau 19) nécessaires à l'administration SC.
3. Administrez les injections sous-cutanées consécutivement, chacune à un site d'injection différent, dans la cuisse, partie supérieure de l'arrière du bras ou l'abdomen,à l'exception d'une zone de 5 cm autour du nombril. La zone de la ceinture doit également être évitée.
4. Lors de l'administration des injections sous-cutanées, il est recommandé d'utiliser différents quadrants de l'abdomen, de la partie supérieure des cuisses ou de la partie supérieure de l'arrière du bras pour espacer chaque injection sous-cutanée de 2,5 ml de casirivimab et d'imdevimab. NE PAS injecter dans zones où la peau est sensible, contusionnée, rouge, dure, ou non intacte.
5. Assurez une surveillance clinique des patients pendant l'administration et observez- les pendant au moins une heure après l'administration.
Elimination
Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
Les aiguilles et les seringues ne doivent jamais être réutilisées.
Placez toutes les aiguilles et seringues usagées dans un conteneur pour objets pointus et tranchants.
Réservé à l’usage hospitalier
Solution à diluer pour perfusion intraveineuse (IV) ou solution pour injection sous-cutanée (SC).
Solution transparente à légèrement opalescente, incolore à jaune pâle avec un pH de 6,0.
Casirivimab et imdevimab sont fournis dans des flacons en verre (type 1) avec un bouchon en caoutchouc et une capsule aluminium amovible.
Chaque emballage contient 2 flacons par boîte :
- 1 flacon de 300 mg/2,5 mL de casirivimab et 1 flacon de 300 mg/2,5 mL d'imdevimab
Chaque flacon unidose de casirivimab de 6 mL contient 300 mg de casirivimab dans 2,5 mL (120 mg/mL) et chaque flacon unidose d'imdevimab de 6 mL contient 300 mg d'imdevimab dans 2,5 mL (120 mg/mL).
Casirivimab et imdevimab sont des anticorps monoclonaux humains d'isotype immunoglobuline humaine G-1 (IgG1)
Pour la liste complète des excipients, voir rubrique Liste des excipients.
L-histidine
monochlorhydrate d'histidine monohydraté
polysorbate 80
saccharose
eau pour préparations injectables